
To Issue 130
Citation: Allmendinger A, Mahler H-C, “How ten23 health is Meeting Developmental Challenges for Intravitreal Products”. ONdrugDelivery, Issue 130 (Mar 2022), pp 43–48.
Andrea Allmendinger and Hanns-Christian Mahler highlight new regulatory and quality requirements for intravitreal applications and discuss technical challenges during drug product technical development and manufacturing.
“The successful administration of IVT injection of typical injection volumes between 25 and 100 μL requires the development of highly concentrated formulations of the desired biologic, which presents major challenges during technical development and manufacturing.”
In an article published in ONdrugDelivery, April 2021 – “State-of-the-art solutions for ophthalmic sterile drug product manufacturing” – swissfillon explained why ophthalmic products require a high level of process knowledge and state-of-the-art manufacturing technologies and expertise to maximise safety, meet stringent regulatory requirements and minimise costs. The article explained how swissfillon’s aseptic manufacturing services, now part of ten23 health, and its innovative highly flexible automated filling line at its facility in Visp, Switzerland, meet the requirements and complexities associated with intravitreal (IVT) and other parenteral preparations. In this article, new regulatory and quality requirements for IVT applications will be discussed, along with technical challenges during drug product technical development and manufacturing.
Retinal disorders include diabetic retinopathy and age-related macular degeneration. They can be treated with anti-VEGF (vascular endothelial growth factor) drugs, which are usually administered by IVT injection into the back of the eye using prefilled syringes (PFSs). This allows direct delivery into the vitreous humour to achieve sustained levels of drug solutions and evade the blood-retinal barrier. IVT preparation requires very high-quality primary packaging and drug product manufacturing technologies to ensure patient safety, regulatory compliance and adequate product design for usability.
The successful administration of IVT injection of typical injection volumes between 25 and 100 μL requires the development of highly concentrated formulations of the desired biologic, which presents major challenges during technical development and manufacturing. Ideally, the drug product is designed to reduce injection frequency and prolong therapeutic levels.
In addition, ready-to-use injections using drug-combination devices, such as PFSs, are preferred, which adds another level of complexity and major challenges to drug product development.
“Silicone droplets may be detected as subvisible particulates or even visible particulates in the product, depending whether the formulation is stored in the silicone-coated syringe and/or in contact with the syringe during administration.”
NEW REGULATORY REQUIREMENTS
The European Pharmacopoeia (Ph Eur) has recently updated its dosage form monograph “Parenteral Preparations”, adding the sub-category of “Intravitreal Preparations”. IVT preparations are defined as “sterile parenteral preparations intended for administration or implantation into the vitreous humour. They are solutions, colloidal dispersions, emulsions, suspensions or implants and comply with the requirements for injections or implants, as appropriate. Unless otherwise justified and authorised, they are supplied in single dose containers and do not contain any preservatives”.
PARTICULATES FOR IVT PREPARATIONS REVISITED
The requirements for IVT preparations in the Ph Eur related to visible particulates (Ph Eur 2.9.20) as well as sub-visible particulates (Ph Eur 2.9.19) aligned with the requirements of other parenteral dosage forms (0520). These are “practically free of particles” for visible particle inspection and a maximum of 6000 particles/container for particle sizes ≥10 μm and a maximum of 600 particles/container for particles ≥25 μm.
This is different from the US Pharmacopeia (USP), chapter <789>, where, for example, significantly tighter limits are defined for IVT preparations, e.g. not more than (NMT) 50 particles/mL for particle sizes ≥10 μm and NMT 5 particles/mL for particles ≥25 μm. Interestingly, there are no controlled clinical studies that relate to the clinical relevance of particulates after IVT injection, although particulates may – or may not – have clinical relevance in the eye, depending on a few parameters, such as their ability to dissolve in the vitreous humour or their density (e.g. if floating).1 Whether any such particulates impact patients’ vision is probably a significant consideration.
“In the case of IVT formulations particularly the choice of excipients is extremely limited, and only a few excipients in sufficiently high concentration have demonstrated clinical safety and regulatory acceptance.”
In general, particles may originate from the formulation in the final primary packaging as a result of protein or excipient degradation, or they may be extrinsically introduced during the manufacturing process.
Syringes are used either as final primary packaging for IVT preparations or for withdrawal and administration when using a vial-based product. Most syringes are siliconised to ensure lubrication and functionality (break loose and gliding forces) for the user. Hence, unsurprisingly, silicone droplets may be detected as subvisible particulates or even visible particulates in the product, depending whether the formulation is stored in the silicone-coated syringe and/or in contact with the syringe during administration.
Subvisible particle measurements, which are traditionally performed with classical light obscuration techniques, would count the droplets as part of the overall particulate product load, and products may fail the tight USP <789> criterion. This is where morphological characterisation tools for particulates, such as flow imaging, add value. These allow particulates to be differentiated qualitatively and provide insights into whether the particulates measured by light obscuration are indeed “contaminants” or “degradants”, or just silicone oil droplets (Figure 1).

Figure 1: Illustrative examples of particles analysed by micro flow imaging extracted from Agra et al.4 High particle variability across siliconised and oil-free syringes and needles from the same lots. A) Silicon oil droplets. B) Other contaminants.
Polymer syringes and other silicone-free syringes may be an alternative to siliconised syringes but must be carefully selected based on drug-product device compatibility and device functionality and syringe quality.
Interestingly, silicone is also used for optical surgery, where approximately 2–3 mL silicone is used in the eye.2 Minute amounts of silicone droplets are therefore typically considered not to be of significant medical concern.3
ENDOTOXIN REQUIREMENTS FOR IVT ADMINISTRATION
Endotoxins are a key consideration for parenteral preparations that relate to patient safety. IVT injections, in particular, require careful consideration for endotoxin limits, as they may relate to possible reversible and dose-dependent inflammation in the eye of different severity and reversibility. Inflammation of the eye may result from acute influx of protein and leukocytes into the anterior and may possibly also manifest clinically as haze persisting in the vitreous and lead to other safety concerns including immune response.
The no-observed-adverse-effect level (NOAEL) in both rabbits5 and non-human primates6 was reported as “not more than (NMT) 0.01 EU/eye.” Endotoxin levels at 0.02 EU/eye and >0.04 EU/eye were found to induce dose-related inflammation.
This NOAEL is especially important considering that host cell lines used for the production of the API may be microbial. In addition, endotoxins may relate to different sources throughout the overall drug substance and drug product production process. The FDA Guidance for Industry (2015) on endotoxin testing recommendations for single-use intraocular ophthalmic devices discusses the limits of NMT 0.2 EU/mL, which, for IVT injection volumes of 10–100 μL, translates to 0.002–0.02 EU/eye, respectively, and, therefore, slightly higher acceptance criteria than the NOAEL derived from preclinical data.
TECHNICAL CHALLENGES OF LOW INJECTION VOLUMES
IVT injections have a defined volume range. The upper volume of IVT injections is typically around 100 μL, governed by medical concerns over clinically relevant symptoms, including an increase in intraocular pressure. The lower limit of IVT injections, typically around 25 μL, is governed by the precision of lowest administration volume and dose, which is influenced by syringe/needle configuration. In some cases, where only parts of the filled product volume are injected, such as 10 μL of drug product, the dose precision may be affected by the user and product usability, with influencing factors, such as graduation marks or the impact of air bubble in the syringe/needle.
It is important to note that product usability and the variability of dosing volume must be established in appropriate studies, including human factors studies. There is anecdotal evidence that (manual) down-dosing for an intended 10 μl injection actually yields doses corresponding to injection of zero μL (air bubble) to 30 μL (3 times the intended dose).
HIGHLY CONCENTRATED FORMULATIONS: VISCOSITY AND INJECTION FORCES
Compatibility with the primary packaging container is typically tested in long-term stability studies as well as in stress studies to test specific liabilities, for example, stability and sensitivity in final primary packaging. When considering plastic (polymer) primary containers, additional parameters are recommended for testing due to the higher permeability of such containers in comparison with glass, including testing for oxygen sensitivity and extractables/leachables.
Given the limited injection volume for IVT administration, the need for high concentration formulations of protein APIs is typically warranted – depending on the target dose. However, protein concentrations correlate exponentially to product viscosity (Figure 2). This means that protein formulations for IVT use are often characterised by high viscosity, which directly implies increased injection forces, as described below.
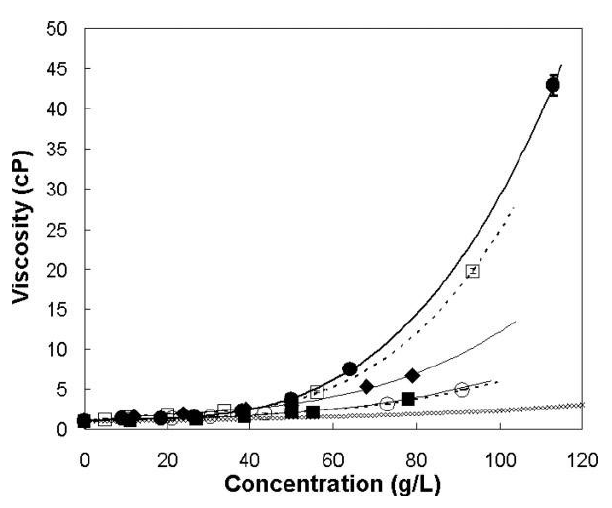
Figure 2: Concentration-dependent viscosity of different antibody solutions at pH 6 and two ionic strengths. (●) 10 mM histidine, (□) 2.2 mM sodium phosphate, (○) 10 mM histidine, 150 mM NaCl, (■) 2.2 mM sodium phosphate, 150 mM NaCl, (◆) 10 mM histidine, 2 mM NaCl, (♢) Viscosity of an equivalent hard-sphere using the Mooney approximation. Lines represent a polynomial fit. Modified according to Salinas et al.7
As some protein formulations can show shear-thinning behaviour facilitating administration, it is imperative to characterise the shear-dependent flow behaviour of such formulations adequately during product development (see Table 1). Besides API concentration, viscosity is also dependent on temperature. Products stored at 2–8°C intended for administration at room temperature typically require equilibration. In the absence of such an equilibration phase, the viscosity at 2–8°C product temperature is likely to be significantly increased and administration potentially compromised.
| Product characteristic: viscosity | Device characteristic: needle inner diameter |
| Characterisation of viscosity-protein concentration dependence | Choice of injection needle depending on product viscosity, user requirements and clinical considerations |
| Characterisation of viscosity-shear-rate dependence assessing (non-) Newtonian behaviour |
Characterisation of variability in needle inner diameter |
| Characterisation of viscosity-temperature profile |
Target setting and failure mode assessment (edge of failure assessments) of injectability (e.g. forces, time) in relation – to intended specification ranges – to user capabilities |
| Choice of formulation (excipients) | |
| Specification setting (content) |
Table 1: Product and device characteristics studied and controlled in development studies to mitigate delivery challenges.
“Filling of low volumes in primary packaging material is technically
challenging, and sterile manufacturing capabilities,
parameters and process set-up dramatically influence the lower fill volume and its related fill precision.”
EXCIPIENTS
To mitigate high viscosity from a formulation perspective, the choice of an adequate formulation is crucial. A common consideration in managing formulation viscosity is the choice of appropriate pH and the use of one or several excipients. Amino acids or salts have been described as potentially managing product viscosity. However, a key parameter for the choice of an appropriate formulation is not only product viscosity but also ensuring that the API is sufficiently stable during manufacturing, transport, (long-term) storage and administration. In the case of IVT formulations particularly, the choice of excipients is extremely limited, and only a few excipients in sufficiently high concentration have demonstrated clinical safety and regulatory acceptance.
DEVICE DESIGN
Ready-to use injections are typically preferred for IVT applications using drug-combination devices, such as PFSs. As most early-stage clinical studies use vials, the ability to switch between product configurations from a vial to a PFS is extremely helpful, allowing the manufacture of both configurations at the same manufacturing facility.
Syringes for ocular administration are typically provided with a flexible Luer-lock system allowing the user (ophthalmologists) to choose the appropriate and preferred needle. The needle needs to be carefully chosen and selected in the context of the product parameters. According to Hagen- Poiseuille’s law, injection forces depend on the inner radius of the needle to the power of 4 – this means that very subtle variations in the inner diameter of the needle lead to significant differences in required injection forces, and hence product usability and performance. Typically, 30G needles are used for IVT injections to minimise corneal damage, which may be caused by larger diameter needles. This provides specific limitations and challenges related to the maximum viscosity of a formulation in the context of a maximum injection force, as defined in user studies.
THE FILL-FINISH PROCESS
Filling of low volumes in primary packaging material is technically challenging, and sterile manufacturing capabilities, parameters and process set-up dramatically influence the lower fill volume and its related fill precision. As an example, gravimetric fill control, as implemented at swissfillon, guarantees a filling accuracy of ±5% and accuracy within 2%, which is suitable for IVT preparations at these very low fill volumes. In addition, swissfillon’s filling line ensures the rejection of single syringes rather than large numbers of syringes, which can result in substantial savings.
Fill volumes must be defined to achieve adequate extractable volumes, and overfills are ideally minimised to save costs. It is therefore recommended that filling parameters are defined in related process studies. In addition, stoppering of low volume dosage forms can provide specific technical challenges, which can be mitigated by the appropriate expertise of a contract development and manufacturing company (CDMO).
Headspace in syringes used for IVT administration is generally undesirable from a clinical perspective. Hence, manufacturing syringes for IVT administration that ensure zero headspace (bubble-free) are desirable. Especially in the case of ophthalmologic products, external sterilisation by ethylene oxide or hydrogen peroxide may be required by specific markets and countries. Residual contaminants need to be specified and their impact needs to be assessed regarding compatibility with the drug product compromising drug substance stability.
THE CONTAINER CLOSURE SYSTEM
A container closure system (CCS) refers to the sum of packaging components that together contain and protect the dosage form. This includes both primary and secondary packaging components, if the latter are intended to provide additional protection to the drug product. The suitability of the CCS for its intended use needs to be demonstrated, including protection of the drug product from, for example, moisture, oxygen or light; compatibility of the packaging components with the drug substance; safety including extractable studies and their toxicological evaluation; and performance, such as functionality and delivery of the intended dose.
An integral part of CCS testing is the evaluation of container closure integrity (CCI), which is an obligatory critical quality attribute for sterile dosage forms. CCI testing evaluates whether there is a leak in the CCS that may allow contaminants (such as microorganisms) from the outside environment to potentially compromise sterility.
USP chapter <1207> (2016) and the revision of the EU Annex 1 for sterile product manufacturing (2020) currently represent the most thorough guidance documents on CCI concepts for sterile injectable products. USP chapter <1207> provides an overview of common CCI tests and categorises them into deterministic (e.g. electrical conductivity and capacitance test, headspace gas analysis, vacuum or pressure decay, and tracer gas vacuum mode, such as helium leak detection by mass spectrometry) and probabilistic tests (such as the conventional blue dye test). Deterministic tests are usually preferred for CCI evaluation.
Additionally, the use of positive and negative controls, including artificial leaks, is key. Importantly, the evaluation of the CCS should not be viewed as limited to end-product quality control testing rather than ensuring that the choice of components is qualified for integrity (CCI) based on quality-by-design, considering variations in primary packaging components but also considering processing. CCI testing strategies remain challenging, dependent on packaging material and configuration and there is no one-size-fits-all CCI testing set-up. Ideally, CDMO partners have established deep expertise and access to a toolbox that may be further optimised for a specific product configuration using science-based and quality-by-design-based approaches for CCS selection, method development and validation.
SUMMARY
In this article, the discussion about IVT products was continued with a key focus on challenges related to product design and development, and updates on any requirements or quality considerations.
Specifically, the following specific requirements and aspects were discussed, which must be considered to manage the complexity related to sterile product manufacturing of IVT injection drug products:
- Most early-stage clinical studies use vials but syringes are a preferred product configuration to facilitate usability. Switching between product configurations to manufacture both vials and PFSs at the same manufacturing facility will be facilitated by a partner with significant expertise to accompany such a switch.
- IVT preparations have specific regulatory requirements related to subvisible particles in the US, and the choice of appropriate methods and characterisation tools is a key consideration.
- IVT injection volumes are low, and individual containers should preferably be filled with only the patient-specific volume (dose) to avoid any down-dosing or product-handling issues. Hence, manufacturing facilities and processes that show highest fill precision accuracy at these very low volumes are key. Minimising over-fill, other than what is required to ensure extractable volume and accurate dosing, can achieve significant cost savings.
- Formulation viscosity and choice and design of formulation are of crucial importance. Identifying the right balance between optimal stability and viscosity, in relation to the choice and characterisation of the devices and needles used for administration is a key success factor.
- The selection of an appropriate CCS is not only a critical priority from a product use and administration perspective but must ensure integrity over its whole shelf life.
A CDMO such as ten23 health is appropriately positioned to support such challenges of IVT injections. In addition to manufacturing complex and high-precision containers in its swissfillon facility, ten23 can assist with formulation services, analytical development and characterisation and process design.
REFERENCES
- Patel S et al, “Evaluation of protein drug performance with vitreous humor in a novel ex-vivo intraocular model”. Eur J Pharm Biopharm, 2015, Vol 95(Pt B), pp 407–417.
- Antoun J, “vitreoretinal surgery with silicone oil tamponade in primary uncomplicated rhegmatogenous retinal detachment”. Retina, 2016, Vol36(10), pp 1906–1912.
- Bakri SJ, Ekdawi NS, “Intravitreal silicon oil usage after intravitreal drug injection”. Retina, 2008, Vol 28, pp 996–1001.
- Agra et al, “High particle variability across siliconized and oil-free syringes and needles from the same lots”. Sci Rep. 2021, Vol 11, p 4645.
- Bantseev V et al, “Determination of a No-Observable Effect Level for Endotoxin Following a Single Intravitreal Administration to Dutch Belted Rabbits”. Invest Ophthalmol Vis Sci, 2017, Vol 58(3), pp 1545–1552.
- Bantseev V et al, “Determination of a No Observable Effect Level for Endotoxin Following a Single Intravitreal Administration to Cynomolgus Monkeys”. J Ocul Pharmacol Ther, 2019, Vol 35(4), pp 245–253.
- Salinas et al, “Understanding and modulating opalescence and viscosity in a monoclonal antibody formulation”. J Pharm Sci, 2010, Vol 99(1), pp 82–93.