To Issue 147
Citation: Asok D, “Strategies for Test Method Validation of Drug Delivery Platforms.” ONdrugDelivery, Issue 147 (May 2023), pp 40–43.
Deepu Asok discusses the importance of test method validation for drug-device combination product development and how a common mistake is to design the validation process for a specific device rather than to accommodate the potential variety of a device platform.
With the recent rise in use of biologics for treating chronic diseases, the relevance and application of injectable drug delivery systems are on the rise. The global injectable drug delivery devices market reached a value of nearly US$39.9 billion (£32.1 billion) in 2022 and is expected to grow to $58.1 billion by 2027 at a rate of 7.5%.1 As more therapies shift towards self-administration in an at-home setting, the need for innovative injection devices is on the rise.
This rising demand has forced both device and drug manufacturers to think in terms of delivery platforms instead of one-off delivery devices. However, this means that manufacturers must become creative when designing their studies to gather evidence about the safety and effectiveness of these delivery platforms. A key element to ensure the success of those studies is the test methods used to verify the safety and efficacy of the devices. The strength of the data rests upon the foundation of the test method validation (TMV), which establishes that the test methods are robust, repeatable and fit for purpose.
WHAT IS TEST METHOD VALIDATION?
When a design is established based on specified design inputs, it becomes the responsibility of the device manufacturer to confirm that the design outputs meet the predefined acceptance criteria. The aim of design verification is to ascertain that design outputs not only fulfil the requirements detailed in the design input document(s) but also evaluate the efficacy of risk controls. Pursuant to this, test methods function as the tools for generating data for design verification tasks. Similar to any tool, the data gathered from them are only as reliable as the tool itself. If a tool is defective, it will produce inaccurate measurements, which, in turn, will lead to incorrect conclusions. Therefore, it is essential to validate test methods to guarantee repeatable and reproducible results; this is vital for the success of any product development project.
A standard TMV process involves designing a study wherein multiple operators conduct a study where they gather test data using the proposed test method. The study will also involve multiple trials on the same (in the case of non-destructive tests) or similar (in the case of destructive tests) test articles. Once gathered, the test results are analysed for variability within the same operator’s data and between that collected by different operators. As long as the total variability falls within the range of the acceptable variability limits previously established, the method is deemed suitable for gathering design verification data to be used in the submission.
Conducting TMV studies for multiple test methods can involve a significant amount of time and resources. The whole process involves significant planning and preparation, including standalone supporting documentation, such as TMV plans, protocols and summary reports. A common mistake lies not in the execution of such studies but rather in not thinking holistically about the larger needs of the organisation and future device platforms.
THINK IN PLATFORMS, NOT PRODUCTS
One of the common mistakes that people make when conducting TMV is that they design their validation plans around the product, not the platform. For example, executing the TMV using a specific prefilled syringe (PFS) or autoinjector design that the developer plans to file for the immediate submission. Then, a few months later, they realise that there is a new PFS design that needs TMV as well. However, it may not be possible to use the previous TMV study, as the validated range is specific to the product that was included in that study and the new product’s performance data falls outside that validated range.
In this case, there is no other choice but to repeat the TMV study for the same test methods, but this time with the new product design. This could require a significant investment in terms of time and effort that could have been saved if the TMV was strategically designed to cover future design variations within that injectable device category. After all, all PFSs must adhere to the same test methods prescribed in ISO standards, such as ISO 11040, 7886 or 11608.
INCLUDE PRODUCT VARIANTS IN THE ORIGINAL TMV STUDY
To avoid this problem of having to repeat TMV studies, try to include multiple product variations within the same study. For example, if there is a 5, 10 and 15 mL version of a syringe, think of ways to design a test method that be used for all these variants. As shown in Figure 1, this can extend the validated range of a test method. Including at least two more product variants, one for a lower data range and one for a higher data range, will establish a validated test method that can be used for multiple design verification studies in the future.
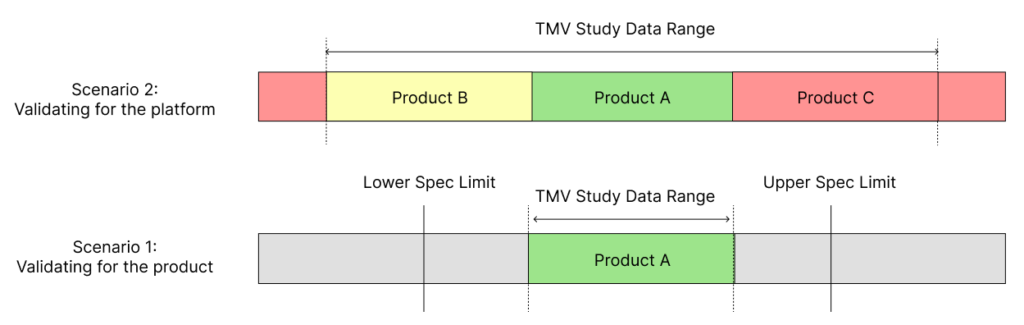
Figure 1: Scenario 1 shows the limitations in the validated data range when using just a single product or part for TMV studies. Scenario 2 shows how using several parts or products within a TMV study provides a wider validated range for the test method.
INCLUDING CHALLENGE PARTS IN TMV STUDIES
As discussed, it would be beneficial to include multiple product variations within the same TMV study to avoid the need for future repetition of work. However, during early studies, it may be challenging to obtain different variations of the injectable device. In such cases, the concept of challenge parts can be invaluable.
Challenge parts are custom-engineered test articles that simulate the form and mechanical function of the product family under investigation. For example, when validating the break-loose and extrusion force of a PFS with only one available variant, an engineered sample can be created to replicate the syringe’s form and function, such as a piston-barrel system. This sample can then be designed to encompass a broader range of break-loose and extrusion forces than the product intended for verification.
By adopting this challenge parts approach, the validated range of the test method can be extended, while also incorporating the final product to be tested during design verification testing in the comprehensive validation study (Figure 2). However, it is important to ensure that the product that will ultimately be tested during design verification testing is included when conducting TMV studies with challenge parts.
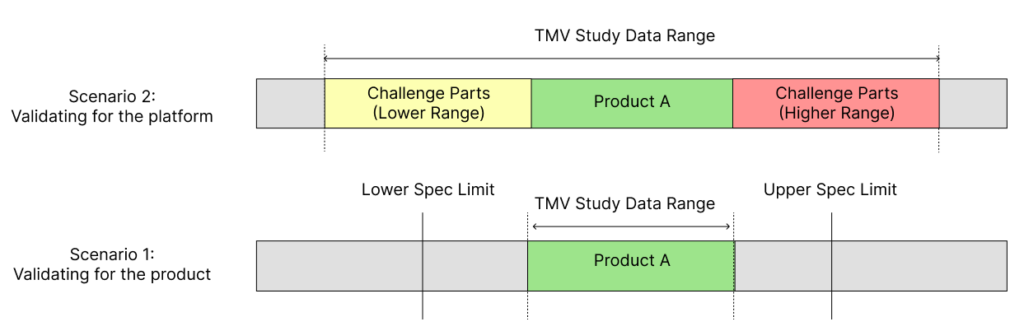
Figure 2: Scenario 1 shows the limitations in the validated data range when using just a single product or part for TMV studies. Scenario 2 shows how using engineered challenge parts within a TMV study provides a wider validated range for the test method.
DESIGNING UNIVERSAL TEST FIXTURES
One of the key elements of a test method is the test fixture that is used to hold or manipulate the test article. Instead of designing a fixture around the specific dimensions of an injectable device, it is valuable to develop a design that can accommodate potential variations of future designs. For example, a PFS can come in different shapes, materials and form factors. It could be one with a Luer or non-Luer tip design, one with a glass flange or an add-on flange adaptor, or one with or without a safety device. It is possible to design a fixture that can be adjusted to accommodate these variations without significantly altering the physics of the testing mechanism.
This can be achieved either by designing fixtures that can be adjusted to accommodate the variations in form factor or by using replaceable adapters that can be swapped in and out based on the changes in device design. For example, one of the most common test methods for PFSs is needle-shield removal force, which is often tested by constraining the syringe body and using a pull fork to pull the needle cap out and measure the force using a universal testing machine. In this case, pull forks can be designed to accommodate the variations in needle shield diameters and use replaceable syringe holder adaptors to include more than one size of syringe in the TMV study.
LEVERAGING EXISTING TEST METHODS
In some scenarios, it may be possible to leverage existing validated test methods instead of starting from scratch. However, it is important to establish a detailed rationale explaining how these test methods are substantively similar. The primary emphasis should be placed on looking at the differences, if any, and explaining how those differences would not impact the validity of the test method. The five key factors that contribute to the repeatability and reproducibility of test methods are equipment, fixtures, procedure, operator and environment. While leveraging previous test methods, make sure to include a side-by-side comparison of these five factors and justify any identified differences that would not impact the test results (Table 1).
| Test Method Attribute | Test Method 1 | Test Method 2 | Rationale for Differences (if any) |
| Equipment | Equipment A | Equipment B | |
| Fixture | Fixture A | Fixture B | |
| Operator | Operator A | Operator B | |
| Procedure | Procedure A | Procedure B | |
| Environment | Environment A | Environment B |
Table 1: Sample table for analysing the differences between the key factors for two test methods.
- Equipment: The measurement equipment used is a crucial element of test method validation. If a different model is used, it is necessary to establish that the new equipment is substantively similar to the original one. For example, Instron (MA, US) and ZwickRoell are two popular makes of equally capable universal testing machines that are used in the industry. By providing a comparison, including a detailed analysis of the two pieces of equipment’s capabilities, operating ranges and precision, it is possible to justify leveraging a TMV study that used one piece of equipment to support testing performed using the other.
- Fixtures: The primary purpose of a test fixture is to constrain the test article in place during the testing process. While using existing test methods, it is important to ensure that there is no significant difference in the fixture mechanism that could impact the results. For example, the repeatability of the test data gathered could vary when holding a syringe using a simple adjustable jaw-type fixture versus a custom-designed syringe-holder fixture.
- Procedure: The test operators must strictly follow the same procedure during TMV and design verification testing. When using existing TMV studies, any deviations from the original procedure must be scrutinised to ensure that they do not impact the validity of the test method. For example, a change in the testing speed for a syringe extrusion force from 100 mm/min to 300 mm/min could produce different test results. However, it does not impact the validity of the test method as a whole as long as the new data is still within the previously validated range.
- Operator: The operator’s level of skill and training play a significant role in any test method. When using a validated test method, it is essential to ensure that the operators will receive substantively equivalent or more training and supervision than the operators involved in the TMV study.
- Environment: Environmental factors, such as temperature, humidity and vibration, can impact test method parameters. Careful analysis of any differences in the environment between the original TMV study and the new study must be carried out to identify any potential impact. For example, a study that was validated in a carefully controlled R&D lab may not produce the same results when performed in a manufacturing environment with extreme environmental conditions.
CONCLUSION
Embracing a strategic approach to TMV by focusing on platforms rather than individual products can significantly enhance the efficiency and adaptability of the validation process. By adopting strategies such as designing universal fixtures, incorporating challenge parts and leveraging existing test methods, manufacturers can reduce the development cycle time for drug delivery devices. This accelerated development ultimately enables them to bring life-changing therapies to patients more quickly, improving patient outcomes and addressing urgent healthcare needs while staying ahead in the rapidly evolving world of injectable drug delivery systems.
REFERENCE
- “Reportlinker.com announces the release of the report: Injectable Drug Delivery Devices Global Market Report 2023”. Press Release, Reportlinker.com, February 10, 2023.