To Issue 160
Citation: Adler M, Reddy Goli VA, Allmendinger A, Mahler H-C, “Transitioning from Vial to Subcutaneous Injection Devices for Biological Drug Products”. ONdrugDelivery, Issue 160 (May 2024), pp 8–14.
Michael Adler, Venkata Appa Reddy Goli, Andrea Allmendinger and Hanns-Christian Mahler, discuss the various factors that need to be considered when transferring to a delivery device for subcutaneous injection, such as a prefilled syringe or autoinjector, during clinical trials or post launch, especially when transferring from a vial format for intravenous infusion.
The subcutaneous (SC) route is becoming increasingly important for the administration of biologics. With the development of SC injection devices, the paradigm of drug product development has continued to shift towards patient centricity by enabling self-administration and ease of use for patients, improving compliance and adherence and reducing the burden on healthcare systems.1 The number of approved monoclonal antibody (mAb) products has been increasing over the last few decades, with a proportional increase in the number of SC products (Figure 1), based on the information compiled by ten23 health on US FDA-approved products.
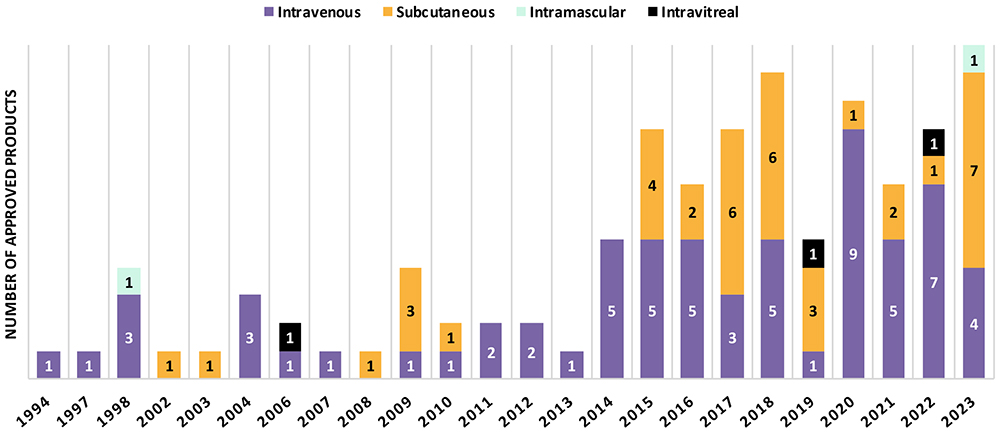
Figure 1: Route of administration of FDA-approved monoclonal antibody products.
“41% of mAb products approved between 2015 and 2023 are administered by SC injection, whereas it was 24% between 1994 and 2014.”
About 79 mAb products, excluding antibody-drug conjugates (ADCs), were approved between 2015 and 2023, which is twice the number of products approved between 1994 and 2014 (31 products). 41% of mAb products approved between 2015 and 2023 are administered by SC injection, whereas it was 24% between 1994 and 2014. In 2023, an impressive number of mAb products (7 out of 12) were approved for SC administration, marking the highest number per year in the past 30 years. Numbers in other countries (e.g. EU approvals) are in line with the trend towards SC administration of biologic injectables.
As innovators change their product lifecycle management approach towards increased patient centricity, transitioning from intravenous (IV) infusion to SC administration with an injection device can be a huge game-changer (patient focus, compliance, differentiation from competitors), with notable examples being Herceptin® (trastuzumab, Roche/Genentech), MabThera® (rituximab, Roche/Genentech), Actemra® (tocilizumab, Roche/Genentech), Benlysta® (belimumab, GSK), Entyvio® (vedolizumab, Takeda) and others. There are also cases where biosimilars have been developed for SC administration when originators were solely available for IV.
Advances in the understanding of SC delivery and SC drug delivery technologies have unlocked the perceived “volume limitation” for SC administration, which have traditionally been assumed to have a maximum 0.5–2 mL injection volume. Much larger injection volumes, even up to 10 mL or more, can now be delivered subcutaneously, with 25 mL being studied in a recently published study in the absence of a permeation enhancer.2
In some cases, co- or subsequent administration of hyaluronidases (enzymes that locally and transiently digest the SC tissue) can facilitate administration of larger injection volumes by reducing SC tissue back pressure.3 Using innovative SC delivery devices, such as on-body injectors (OBIs), including West Pharmaceutical Services‘ SmartDose or Ypsomed’s YpsoDose, can also facilitate larger-volume SC injections without a permeation enhancing enzyme. Herceptin Hylecta™ (trastuzumab/hyaluronidase, Roche/Genentech), Rituxan Hycela® (rituximab hyaluronidase, Roche/Genentech), Darzalex Faspro® (daratumumab/hyaluronidase, Janssen) and Phesgo® (pertuzumab/trastuzumab/hyaluronidase, Roche/Genentech) are examples of SC administered drugs exceeding 2 mL using hyaluronidase co-formulations. On the other hand, Repatha® (evolocumab, Amgen) used the Pushtronex® system (West’s SmartDose®) and Ultomiris® (ravulizumab, Alexion) also used SmartDose® to deliver 3.5 mL of solution.
In first-in-human (FIH) clinical trials, biologics are usually administered by IV injection or infusion for various reasons, such as ensuring 100% bioavailability. Some companies already test SC delivery in FIH studies to establish SC injection early on, which saves time for non-clinical and clinical bridging later. Indeed, establishing and understanding bioavailability early during development can be essential to avoid later undesirable surprises, because bioavailability after SC administration may be low due to SC matrix and tissue interaction, depending on the molecular properties of the drug.
Vial presentations often serve the need for early clinical studies, as the dose volume can be easily adjusted for dose escalation and the development of vial presentations is less complex than for injection devices, making it less prone to technical issues that might lead to a delay of FIH clinical trials. The SC administration across different dose groups in the FIH study out of a vial would be achieved by administering:
- Different volumes, such as using disposable syringes with or without SC clinical pumps
- Using a placebo/diluent formulation to dilute to the respectively required concentrations and then administering similar volume, such as by using a disposable syringe with or without SC clinical pumps.
Hence, having a high-concentration formulation for SC administration is highly recommended when choosing a vial presentation for FIH clinical trials. If using a prefilled syringe (PFS) for SC administration in FIH studies, it should preferably contain the highest dose, using either additional dose strengths (concentrations or volumes) or “down dosing” the syringe to administer lower doses (although this approach is not recommended).
Using vials for SC administration in commercial settings may also be a consideration. However, it is obvious that errors and issues with the usability of such products, such as handling vials and solution withdrawal, may present significant issues, depending on the user group. Therefore, a transition from a vial to an SC injection device is common for biologics and can occur either during clinical development or post-launch. These devices include PFSs, autoinjectors, pen injectors and OBIs.
When switching to an SC injection device, either during or after clinical trials, the following scenarios are possible:
- IV vial to SC injection device during clinical development
- IV vial to SC injection device post-launch
- SC vial to SC injection device during clinical development
- SC vial to SC injection device post-launch
- SC injection device to SC injection device post-launch.
Moving from IV to SC (cases 1 and 2) is more complex as it includes a change of the route of administration (RoA). As the bioavailability after SC injection for biologics is less than 100%, implying a modified pharmacokinetic (PK) profile, the dose must be adapted. Knowledge about the anticipated bioavailability is therefore key. Furthermore, some drugs may have other safety and/or efficacy profiles with SC vs IV administration, depending on their mode of action. In addition, because of the smaller injection volume for SC administration compared with IV infusion, a change of formulation and container closure system (CCS) is usually required, necessitating a change in the manufacturing process.
The transition from an IV infusion or SC vial to an SC injection device requires several chemistry, manufacturing and controls (CMC) development activities, as well as clinical and non-clinical, depending on the timing and scope of the change:
- Formulation development
- Primary packaging selection and integration with formulation
- Device selection and device development
- Drug-device integration
- Drug product manufacturing process development
- Bridging studies
– Comparative stability studies, including real-time and accelerated stress stability and characterisation
– Non-clinical studies
– Clinical studies.
CONSIDERATIONS FOR SC FORMULATION DEVELOPMENT
API Concentration
The increasing trend of SC administration has been accompanied by a rise in high-concentration formulations, due to the limited injection volume for SC administration. Since 2015, a notable number of high-concentration (>100 mg/mL) formulations have been approved (44%), with even higher numbers in 2023 (more than 50%). However, the use of large-volume injection products at mid-range concentrations is also on the rise, reducing some of the technical challenges related to high-concentration formulations. Generally, the higher the concentration, the greater the protein aggregation, the higher the viscosity and the higher the concentration of potential impurities from the drug substance process (e.g. lipases), which may also impact stability. Therefore, the concentration of the API is usually a compromise between injectability, stability, manufacturability and acceptable dose volume to achieve the desired dose (Figure 2).
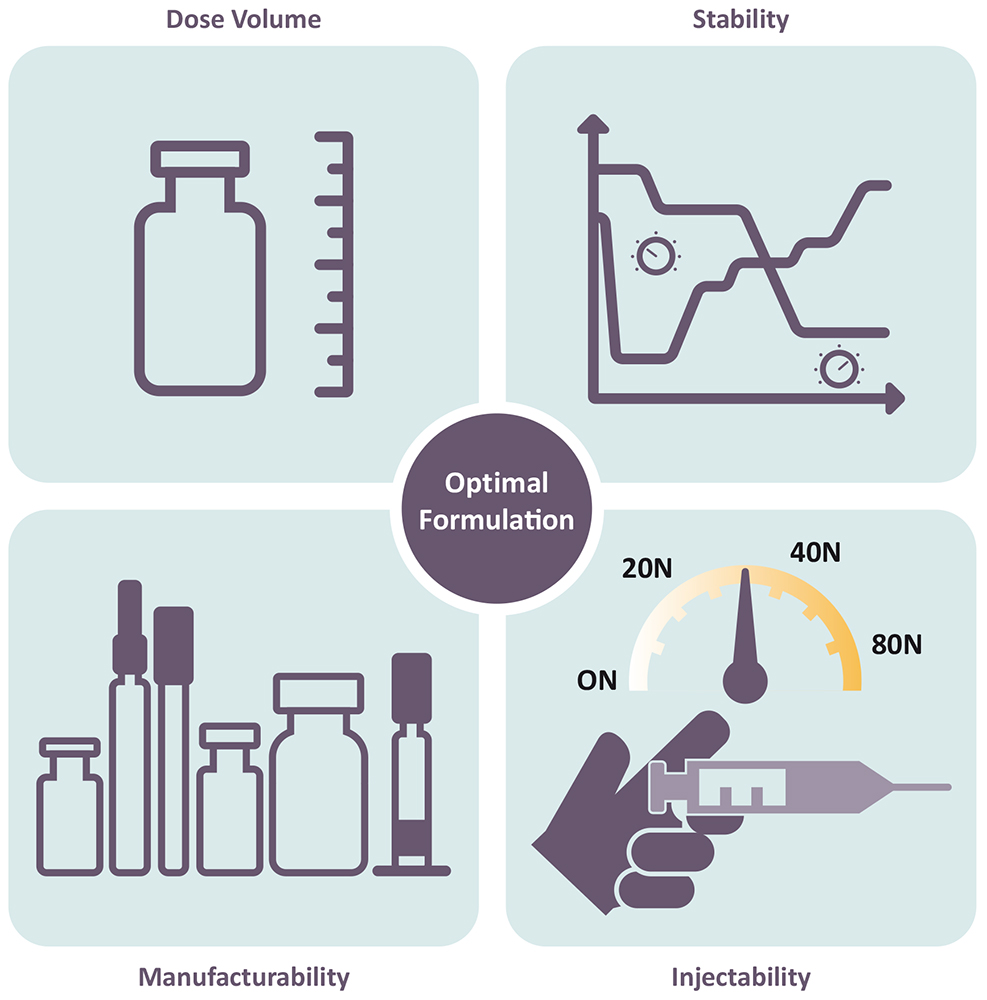
Figure 2: Interplay of critical parameters impacted by API concentration and formulation.
Selection of Excipients
Biologic formulations commonly contain a buffer system (e.g. histidine, acetate) to ensure adequate pH, a stabiliser/tonicity adjuster (e.g. sucrose, trehalose) and a surfactant (e.g. polysorbate 80, polysorbate 20 or poloxamer 188) to protect the protein against adsorption and interfacial stress. Excipients must be safe in the concentrations and doses to be administered (considering the target indications, for example, adult versus pediatric use) and acceptable for parenteral use from a safety and regulatory standpoint.
High-concentration mAb formulations frequently contain excipients such as salts or amino acids to modify or weaken protein-protein interactions, thereby lowering the viscosity while maintaining colloidal stability. Based on ten23 health’s assessments of marketed products, 26 of 48 (~55%) approved high-concentration injectable mAb products contain amino acids, including arginine, proline, glycine and methionine. Some of the high-concentration formulations also use amino acids or chelators to prevent oxidation of the API or polysorbate.
The optimal formulation pH depends on the stability and viscosity, considering tolerability in the patient population. According to ten23 health’s assessment, most SC products are formulated to between pH 5.2 and 6.2, where most of the products show acceptable stability. Histidine remains the leading buffer of choice (~60%) for high-concentration mAb products, followed by acetate. Over the years, the usage of citrate as a buffer has reduced considerably for SC administration compared with the IV route, based on some (poorly studied, in the authors’ view) concerns on citrate formulation causing injection pain upon SC injection. Some products initially approved with citrate buffer were re-formulated, such as adalimumab and ixekizumab. Such re-formulations may also generate intellectual property.
THE EVOLUTION OF SC INJECTION DEVICES
Injection devices represent the pinnacle of convenience for patients, enhancing comfort, adherence and overall satisfaction. These devices, typically arriving as ready-to-use products, streamline handling, eliminating the need for additional manipulation and reducing errors. Specifically designed for SC administration, they empower self-administration, facilitating the transition to at-home care settings and yielding significant economic savings. The engineering behind these devices prioritises patient needs, informed by human factors studies, to ensure optimal usability.
While traditional PFSs and autoinjectors paved the way, recent advancements include larger-volume OBIs and connected devices that enhance treatment scheduling and outcomes. This diversity in product presentation enables flexible care settings, promoting medical compliance. However, the development of injection devices presents additional technical and regulatory challenges compared with vial presentations, requiring careful consideration of drug-device integration and manufacturing, including selection of the primary packaging and device itself. At present, vial presentations remain favoured for early-stage development, offering flexible dosing and fewer technical hurdles, with the transition to devices necessitating further consideration deeper into development. Preferably, such vial to PFS changes would not require any facility or provider tech transfers, which always add additional risk and cost, rather than being manufactured at the same line, facility and vendor, such as ten23 health.
SC INJECTION DEVICE SELECTION
When selecting an SC injection device, several critical factors must be considered to ensure optimal device performance, as well as patient safety and comfort, while also maintaining drug product stability and compatibility with the primary packaging and injection device. Firstly, the dose volume plays a pivotal role in device selection as different devices, such as PFSs, autoinjectors and OBIs, are designed to accommodate varying volumes of medication. Given that the final dose will typically be determined during clinical studies, multiple drug product presentations and strengths may be considered when developing a clinical product for SC administration.
Product viscosity and flow characteristics, such as non-Newtonian behaviour, is another crucial factor as it directly impacts injection force, affecting the injection time dependent on needle size. According to the Hagen-Poiseuille law, a change in the inner needle diameter impacts injection time by the power of four. Thus, device components must be adequately selected and controlled to meet end-user or device requirements. The variability of viscosity, due to the API concentration, and needle inner diameter will determine the failure criteria to be assessed.
Further considerations when selecting the injection device include the selection of the primary packaging container materials, usually glass (siliconised or nonsiliconised) or polymer (e.g. cyclic-olefin polymer), along with rubber (siliconised or non-siliconised) for the stopper. Material choice can impact factors such as gas permeability, which is critical for oxygen-sensitive molecules or preserved multi-use formulations where some preservatives may evaporate across plastics; device performance, including the break-loose and glide force; manufacturability; and product compatibility and stability.
In general, material variability, such as its dimensions, must be specified and controlled. This is especially important in the context of ready-to-use (RTU) components. Factors such as particle contamination, scratches and endotoxins can all be influenced by how RTU components are produced, assembled, stored and shipped, and must be tightly controlled in order to meet requirements. Therefore, to ensure quality, the specifications for such factors should be much tighter for RTU primary packaging than those for the final product’s specifications.
DRUG-DEVICE INTEGRATION
Components to consider when developing an SC injection device are the API, the formulation, the primary packaging/ container closure system and the device. Their characteristics, interplay and manufacturing process may impact:
- Performance (e.g. usability, functionality, injectability)
- Protection of the product against microbial ingress
- Protection of the product against permeation of gases
- Stability/compatibility
- Safety
- Manufacturability
- Transportability.
Several technical development studies are required to address potential risks during drug-device integration (Table 1). Undertaking this process is key to optimising the design of all the components of the injection device and manufacturing process.
| Potential risk | Contributing risk factors | Potential risk mitigations |
| Increase of extrusion force or injection time over shelf life due to silicone-oil migration |
|
|
| Needle clogging due to water vapour permeation through rigid needle shield |
|
|
| Tungsten-induced protein aggregation |
|
|
| Silicone-oil-induced protein aggregation |
|
|
| Leachables |
|
|
| Loss of sterility due to stopper movement during air transport |
|
|
Table 1: Examples of potential risks during drug-device integration, contributing risk factors and potential mitigations.
MANUFACTURING PROCESS DEVELOPMENT
SC formulations are often high- concentration protein formulations, which have a higher viscosity than water or placebo. Highly viscous formulations can impact the drug product manufacturing process. There are a number of aspects that are especially important to consider for manufacturing process development when working with high-concentration formulations for PFSs and cartridges for autoinjectors, pen injectors or OBIs.
Filtration Process
For aseptic manufacturing, formulations must be filtered to achieve sterility. Hence, the ability to be filtered is a key criterion when selecting a formulation. In addition, due to the high costs of the API, especially in high-concentration protein formulations, it is important to minimise losses during filtration. These are driven by:
- The flush volume at the beginning of the filtration process
- The filter hold-up volume.
The flush volume is required to account for binding or adsorption of the API or surfactant to the filter, as well as to the residual water in the filter after the pre-use filter integrity test. Both can result in “underdosing” of the first-filled containers. Filter binding or adsorption for a given formulation is highly dependent on the filter membrane material and type. Therefore, to minimise the losses related to filter binding and adsorption, the filter membrane material should be selected carefully. Also, as filter binding and adsorption is formulation specific, the flush volume needs to be determined, unless it can be defined based on prior knowledge. To minimise the losses related to the hold-up volume, the filter size or effective filtration area should be as small as possible while ensuring a high enough filtration flux and avoiding filter fouling.
Filling Process
Existing filling technologies have their own advantages and disadvantages, such as their accuracy and precision and their potential to induce protein particle formation. As such, selecting a filling technology that is suitable for a given formulation is critical. The filling parameters, including pump speed and tubing diameters, need to be optimised.
The pump speed needs to be synchronised with the motion of the filling needle to avoid dripping, clogging, splashing and foaming, as well as to ensure filling accuracy and precision. Filling needle clogging can occur during filling interruptions, especially when dealing with high-concentration protein formulations. Non-optimised filling parameters can lead to losses and a high reject rate during visual inspection due to a high fill-weight variability or filling needle clogging, which can potentially lead to abortion of the filling process and the need to discard the remaining bulk solution.
Stoppering Process
Stoppering of PFSs and cartridges can be done either by vent tube or vacuum stoppering. Each of the stoppering technologies has its own advantages and disadvantages. Vent tube stoppering generally has a higher throughput than vacuum stoppering, however, some coated stoppers can exhibit wrinkles in the coating after vent tube stoppering due to the compression in the vent tube and later relaxation. As these wrinkles can have an adverse impact on container closure integrity, the potential formation of wrinkles needs to be carefully assessed.
For some SC injection devices, bubble-free filling is required, which can be achieved with adequate knowledge and expertise, such as by using specific vacuum stoppering processes. The selection of the stoppering process also depends on the material properties of the plunger stopper. For example, some plunger stoppers cannot withstand the compression in a vent tube and need to be stoppered by vacuum stoppering.
The stopper position in the container will be based on the fill volume and its tolerances, as well as the process capabilities. Furthermore, it is important to consider the impact of the stopper position on the assembly process and potential stopper movement during transportation, especially by air, where stoppers may move into non-sterile areas.
BRIDGING STUDIES
The extent and scope of a bridging programme is determined by the timing of change – development versus commercial – and the extent of the change between formats. Transitioning from a vial for IV infusion to an injection device for SC injection is the most complex scenario, and typically requires the most extensive bridging studies. Switching from an IV vial to an SC injection device post-launch has the advantage that efficacy, safety and required dose have already been established, and clinical studies may require fewer patients and time if regulators accept a non-inferiority PK study design.
Technical Bridging Studies
Transitioning from an IV vial to an SC injection device usually involves a change of the formulation to one with a higher API concentration to account for the lower bioavailability and limited volume of SC delivery. In some cases, it also involves a change from a lyophilised formulation to a typically less stable high-concentration liquid formulation. To manage stability and viscosity challenges, new excipients might be needed for the SC formulation. Therefore, a technical study is required, which typically includes a comparative stability study comparing pre- and post-change drug product formulations and presentations, for example, a vial and a PFS.
Given the change of the formulation and potentially the dosage form, the post-change drug product is typically expected to be less stable, given the higher concentration of API usually involved. In any case, it is important to assess the overall exposure of any degradant – considering the anticipated dose against the concentration of the degradant – and to assess whether any new degradants are present in the post-change formulation, which may require new toxicological assessments.
Preclinical Bridging Studies
Prior to introducing the SC formulation into clinical studies, at a minimum, local tolerance must be established in a good laboratory practice toxicology study, as per Organisation for Economic Co-operation and Development (OECD) requirements. Additionally, PK and bioavailability may need to be estimated in a suitable animal model.
Clinical Bridging Studies
Prior to introducing the SC formulation into clinical trials, the new formulation at the higher API concentration is typically tested in human PK and bioequivalence studies. Based on the outcome, the dose, injection volume and dosing frequency are defined for later clinical studies.
It should be noted that the SC formulation for the preclinical and clinical bridging studies can theoretically be supplied in a vial presentation and does not have to be supplied in the commercial SC injection device, assuming that the stability of the SC formulation in the vial is comparable to the stability in a syringe or cartridge configuration. Although not generally recommended, Phase III trials can be initiated with an SC vial presentation with an introduction of the SC injection device later, such as in an open-label extension. It is recommended that such study designs are carefully discussed and kept in alignment with the respective regulatory authority.
WHY CHOOSE TEN23 HEALTH?
A CDMO such as ten23 health is appropriately positioned to support its customers with technical and regulatory experience when transitioning from a vial presentation to an injection device during clinical development, as well as post-launch. ten23 health’s team of experts can design an adequate strategy from early stage to commercialisation to de-risk the development approach. ten23 health offers end-to-end services for sterile drug products, including IV and SC formulation development, drug-device selection, integration and testing, manufacturing process development, comparability studies, analytical development, clinical and commercial GMP fill-finish and quality control release and stability testing.
ten23 health provides its GMP fill-finish of complex and high-precision containers at its facility in Visp (Switzerland), including syringes, vials and cartridges, including the capacity to handle both glass and polymer containers on the same line. Therefore, when changing from a vial to syringe or cartridge configuration, no transfers to other lines or facilities are required, making such changes – embedded into their respective technical evaluations – seamless.
For more information, visit ten23 health’s website: www.ten23.health.
REFERENCES
- Allmendinger A, “Opportunities in an Evolving Pharmaceutical Development Landscape: Product Differentiation of Biopharmaceutical Drug Products”. Pharm Res, 2021, bol 38(5), pp 739–757.
- Dang X et al, “Clinical Investigation of Large Volume Subcutaneous Delivery up to 25 mL for Lean and Non-Lean Subjects”. Pharm Res, 2024, Vol 41(4), pp 751–763.
- Bookbinder LH et al, “A recombinant human enzyme for enhanced interstitial transport of therapeutics”. J Control Release, 2006, Vol 114(2), pp 230–241.