To Issue 131
Citation: Lacombe J, Suman JD, “Using Quality by Design to Optimise the Combination Product Journey”. ONdrugDelivery, Issue 131 (Apr 2022), pp 66–69.
Justin Lacombe and Julie D Suman explain how applying quality by design principles to combination products improves development efficiency, aligns with design controls, and maximises product safety and efficacy.
With the shift from quality by test (QbT) to quality by design (QbD) principles in drug development and manufacturing, drug and device developers have adopted a flexible approach that allows them to determine the root cause of a quality issue more quickly and adjust accordingly. Applying QbD principles to combination products is especially valuable due to the complexity inherent in both clinical and commercial manufacturing. Following a QbD approach helps developers identify quality issues earlier in the development process, which helps streamline the path through clinical trials to regulatory approval and commercial release.
WHAT IS QUALITY BY DESIGN?
“A methodical, focused approach is critical for developing products that are consistently safe and effective for their intended patients.”
As applied to pharmaceutical development and manufacturing, QbD refers to a systematic approach that is based on predefined objectives, emphasises product and process understanding and control, and is based on sound science and quality risk management. QbD has been part of ICH guidance since 2005 for drug product development, and has since been extended to include drug substance and analytical method development.
As outlined by the ICH guidance, QbD includes the following elements:
- A thorough identification of the definition of a product through its critical quality attributes (CQAs)
- A systematic evaluation, understanding and refining of the formulation and manufacturing process, including:
– Identifying (through, for example, prior knowledge, experimentation and risk assessment) the material attributes and process parameters that can have an effect on CQAs
– Determining the functional relationships that link material attributes and process parameters to product CQAs - Using the enhanced product and process understanding in combination with quality risk management to establish an appropriate control strategy which can, for example, include a proposal for a design space(s) and/or real-time release testing.
QbD combines the target product profile with quality requirements to arrive at a quality target product profile (QTPP). The objective is to bring together ICH quality risk management, regulatory requirements and design of experiments to arrive at robust products and processes, with the end goal of more efficient design, development and manufacturing.
WHY TAKE A QUALITY BY DESIGN APPROACH?
Combination products, including prefilled syringes, autoinjectors, infusion systems and dry powder inhalers (DPIs) have advanced dramatically over the past several years. Within this context, DPIs – as systems to deliver drugs via inhalation to the lungs – require a deep understanding of the complex formulation-device-patient interplay.
A methodical, focused approach is critical for developing products that are consistently safe and effective for their intended patients. DPIs, for example, require the formulation and device to work in concert to deliver the inhaled dose. Development for DPIs and other combination products includes the integration of QbD principles with process development, design controls and human factors (HF).
Because QbD is a systematic approach that builds quality into product design and development, it enhances development capability and speed, as well as formulation design. As a proactive rather than reactive process, QbD allows product developers to identify issues earlier than they could under a QbT approach, increasing efficiency by focusing on the most important interactions – a priority for all stages of combination product development and manufacturing. QbD also benefits later stages of development as, with an improved understanding of product and process, manufacturers can scale up more easily and efficiently.1
However, successful combination product development and manufacturing using a QbD approach requires varying and advanced expertise. It brings together pharmaceutical science and regulatory disciplines, all related to both drug formulation and device performance, to optimise drug delivery and achieve the best outcome for a development programme.
QUALITY BY DESIGN AND GMP COMPLIANCE
QbD for combination products aligns with good manufacturing practice (GMP) requirements for drug products, as well as the following medical device GMP requirements per 21 CFR Part 820:
- Management responsibility: senior-level oversight of the process from development through to validation
- Corrective and preventative action (CAPA): procedures for implementing CAPAs
- Purchasing controls: quality agreement between supplier and sponsor with well-defined change controls
- Design controls: process for confirming that there is no negative interaction between the drug and device.
DESIGN CONTROL: BRINGING DRUG AND DEVICE TOGETHER
The design and development of a medical device or its constituent parts is performed under the rigorous structured framework of the design controls process. The process involves several steps, starting with identifying patient needs, which is arguably the most important step in the process.
Once the patient needs have been identified, the process continues by translating these needs into design and engineering requirements. These requirements feed into the design process to create a prototype with controlled specifications (i.e. outputs).
Next, developers determine whether the product meets CQA and other design aspects through a verification process, often carried out with support from a device supplier. The finished combination product then goes through a rigorous validation process that includes clinical trials and HF (summative) studies. The primary question to answer during these studies is “Does this combination product address the patient needs set out in step one?”
Following a QbD process aligns well with the medical device design controls process. This, in turn, allows more development activities to occur in parallel than under QbT. Risk management, which takes place throughout the development process, can then encompass and inform both the drug and device development processes (Figure 1).
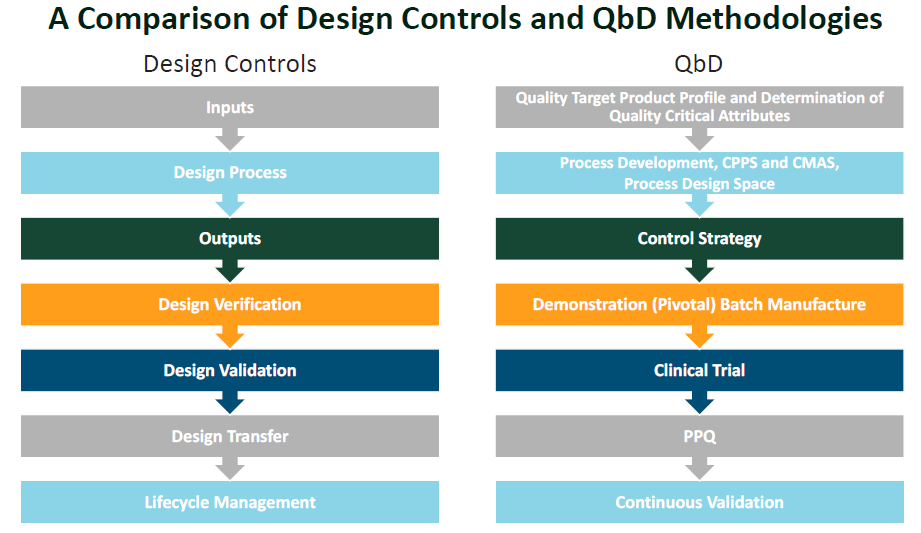
Figure 1: A comparison of traditional and QbD design controls.
“Combination product developers must prove that all the tasks involved in a combination product allow patients to use the product safely and effectively.”
HUMAN FACTORS: A STUDY IN PERFORMANCE
HF studies relate to safety and efficacy but primarily focus on how the patient (or user) interacts with the combination product. HF studies help sponsors uncover the cause of issues that arise during use. Over the past five years, the US FDA has taken a stronger interest in HF studies, issuing guidance on HF information for combination products in 2016.
When conducting HF testing for combination products, defining the patient and patient needs is critical. The patient’s age; whether they will use the product at home, in the doctor’s office or the emergency room; and whether the patient, a caregiver or a physician will administer the product all play a role in HF study design.
HF also considers the patient experience, which differs from traditional oral drug products. Rather than simply swallowing a pill, tablet or liquid, the patient performs several tasks to administer the product – for example, inserting a cartridge, removing a cap, turning on a device, pressing a button, stopping delivery and so on. DPIs that incorporate digital technology (e.g. smart inhalers) may be tailored to patients’ breathing patterns and add additional features to address handling errors.
Failure to correctly perform any of the tasks necessary to administer the drug increases the risk that the patient will not receive the intended dose. Combination product developers must therefore prove that all the tasks involved in a combination product allow patients to use the product safely and effectively.
“Applying QbD principles to combination products not only improves efficiency by identifying quality issues early, it also helps maximise product safety and efficacy, and aligns it better with design controls.”
POINTS TO CONSIDER BEFORE MEETING WITH A CDMO
Clinical and commercial manufacturing of a combination product involves multiple interrelated steps. In addition to formulation, filling and testing, manufacturing these products involves component design, assembly and labelling. These processes interact with one another, multiplying the complexity. When meeting with a contract design and manufacturing organisation (CDMO) for the first time, keep these complexities in mind. Any outsourcing of manufacturing or certain processes to a moulder, product design firm, analytical laboratory or other third-party vendor also increases the complexity of managing the project.
When using a QbD approach, keep the following practices in mind for a more efficient sponsor-CDMO relationship:
Determine Responsibilities
Which part of the assembly process will the CDMO handle versus an outside vendor? Using CDMO-approved vendors instead of a sponsor’s own may require more extensive release testing.
Prepare for Additional Labelling Requirements
When moving a combination product through clinical trials, sponsors may have more extensive labelling requirements. A multidose device, for example, may include labelling for the device itself, the device packaging and the carton it’s transported in.
Define Process Requirements
Manufacturers must determine processes for formulations, filling, assembly, labelling, sampling and inspection. If filling takes place at a different site, sponsors will need to think about shipping steps, storage containers, packaging conditions and allowable hold times. With moisture-sensitive or potent materials, sponsors will also need to consider what part of these processes need to occur within these protected environments. Do operators need protection from highly potent materials? What parts of the system need low relative humidity?
Establish Formulation and Filling Processes
For combination products, manufacturers will need to know both occupational exposure limits and permitted daily exposure limits, as well as formulation method and parameters, and filling mechanisms and limits. Manufacturers also need to be aware of interactions between performance and filling requirements.
Understand Component and Sub-Assembly Requirements and Specifications
Sponsors must prepare well-defined documentation requirements that include all relevant certificates and inspections. Where applicable, include Transmissible Spongiform Encephalopathy and Bovine Serum Albumin certificates. Sponsors will also need to know the function of each component within the combination product and its level of risk. They must also outline and understand which components the CDMO will want to inspect.
Plan for Analytical Testing Requirements
A combination product like a DPI requires extensive testing as part of the QbD process. This includes aerosol performance, microbiological testing, leachables, and physical and chemical attributes. Sponsors should allocate sufficient material to meet the testing and stability requirements for these drug products.
THE MEETING: A RISK-BASED APPROACH TO PROCESS DEVELOPMENT FOR SPONSORS AND CDMOS
Meeting with a CDMO for the first time requires learning and patience on both sides. When preparing to manufacture a combination product, share all processes, requirements and specifications necessary so the CDMO can manufacture the product with consistency and accuracy.
Process development is one of the more involved elements of the CDMO-sponsor relationship. In terms of mitigating risk, process development informed by risk analysis and QbD is one methodology sponsors can use to determine risks involved in certain parts of the manufacturing and development processes. The more information obtained during process development, the better the yields and the higher the quality that can be achieved. As the product moves through development, sponsors will find fewer unknown quality issues during GMP manufacturing.
Applying QbD to process development requires the establishment of an acceptable quality target product profile (QTPP). Establishing a QTTP is achieved by defining product attributes and patient needs(CQAs). These needs are mapped to critical material attributes (CMAs), evaluating the quality impact of critical process parameters (CPPs), and evaluating both metrics together. Risk analysis occurs during process development. Using a QbD approach, sponsors should consider using a risk analysis tool such as failure mode and effects analysis in conjunction with a decision tree to identify the highest risks.
BRINGING IT ALL TOGETHER
Applying QbD principles to combination products not only improves efficiency by identifying quality issues early, it also helps maximise product safety and efficacy, and aligns it better with design controls. Now that regulatory agencies encourage a QbD approach, it’s more important than ever to build quality into the process. Connect QbD principles with design control and HF testing to arrive at combination products that bring the most benefit possible to the patients who need them.
REFERENCE
- Yu L et al, “Understanding pharmaceutical quality by design”. AAPS J, 2014, Vol 16(4), pp 771–783.