
To Issue 158
Citation: Budd G, Suman J, “Advancing OINDP Clinical Trials with Speed and Efficiency: Strategies for Success”. ONdrugDelivery, Issue 158 (Apr 2024), pp 42–46.
Gemma Budd and Julie Suman, consider the key issues that preclinical and clinical trials for orally inhaled and nasal drug products must address within the context of the information that regulators are looking for, providing insight, strategies and advice for both novel and generic products in this sector.
“The preclinical stage of OINDP development is a lengthy iterative process that establishes whether or not a drug is worth taking from discovery into human trials and if it is safe to do so.”
OINDP REGULATORY PATHWAYS
As inhaled and nasal drug delivery becomes increasingly popular, more drug developers are facing the challenge of taking complex orally inhaled and nasal drug products (OINDPs) through clinical trials. This is no easy task, especially with the relevant expertise and capabilities in relatively short supply. Understanding informational requirements and how best to invest scarce resources to de-risk and accelerate progress is vital.
To set the informational requirements for preclinical and clinical trials in context, it is worth considering the multiple regulatory pathways by which OINDPs currently proceed to market. NDAs for small molecules are approved via the 505(b)(1) route – an arduous, high-risk pathway that calls for full preclinical and clinical studies. Biologics are a growing area of research activity, and the biologics licence application (BLA) brings additional requirements for such products.
The NDA 505(b)(2) route, or “hybrid application” in EMA terms, is for new formulations of existing drugs and streamlines progress to market on the merits of existing data. This is the route pursued for drug repositioning, which is a notable trend in the OINDP space. Generic OINDPs are approved via the abbreviated NDA (ANDA) 505(j) pathway, with the most popular targeting localised action in the nose and lung. Approval for a prescription to over the counter (OTC) switch can also be obtained via this route.
These routes are all associated with slightly different requirements when it comes to preclinical and clinical studies. However, there are fundamental similarities and overlaps, notably with respect to chemistry, manufacturing and controls (CMC).
PRECLINICAL: ESTABLISHING A ROBUST FOUNDATION FOR CLINICAL TRIALS
The preclinical stage of OINDP development is a lengthy iterative process that establishes whether or not a drug is worth taking from discovery into human trials and if it is safe to do so. Therefore, the primary goals are to evaluate clinical efficacy realistically and rigorously assess safety.
The key questions to answer with respect to establishing clinical efficacy include:
- How will the drug achieve efficacy?
- Is the intention for local or systemic action and how will the drug reach the target site?
- Will the delivery system significantly influence drug product performance – during in vitro testing, in animal studies and, ultimately, in human use?
- What devices should be used for animal testing, and can they be used for human trials?
- How can animal testing be made as representative as possible?
With respect to safety and toxicology, there is a need to establish the limits associated with use of the drug substance, as well as for any functional excipients being used for the first time or at levels beyond approved limits. Toxicology studies call for the capability to confidently define the identity, strength, purity and quality of functional drug product constituents, taking account of the potential for degradation and recognising that – for OINDPs – the strength of the formulation on the shelf is not the same as what is delivered. Put simply, these studies are about demonstrating the ability to confirm that the chosen analytical and manufacturing processes can securely deliver a drug product of suitable quality for clinical trials and, ultimately, commercial use (Box 1).
BOX 1: ACCELERATING AND DE-RISKING PRECLINICAL TRIALS
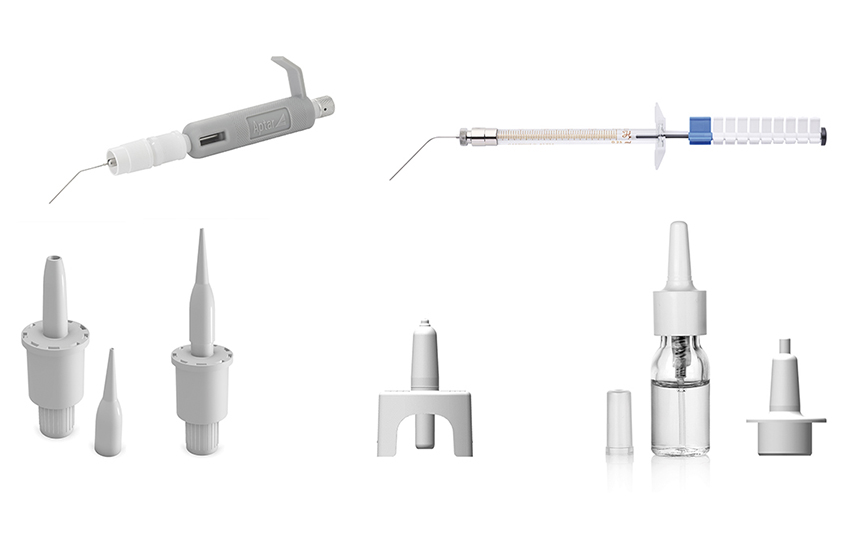
Figure 1: Aptar Pharma’s devices suitable for animal dosing include the PADA and fine mist sprayer (top) for mice, the Unidose (UDS) liquid and powder paediatric (bottom left and middle) and the VP7-232NE paediatric, a multidose nasal spray (bottom right).
“At the preclinical stage, stability requirements only extend to ensuring the consistency of the product for the duration of clinical trials, which could potentially be just a few months.”
PRECLINICAL: PRIORITIES AND PITFALLS
At the preclinical stage, animal testing and product development – including formulation and device characterisation – proceed in tandem, with both presenting the potential for difficulties.
Animal testing with OINDP devices is made complicated by both the inability to control the “technique” of the animal and the species-dependent geometries of the nose and lung. Using specific device actuators for animals and paediatric devices can help but, even with robust data, translation to performance in humans is relatively poor with few reliable models.
This intensifies the need for physiologically based pharmacokinetic(PBPK) models and robust deposition studies, both of which can help to elucidate performance. The timing of animal studies is crucial as, although simple, early formulations (drug and buffer) may be inadequate for proof of- concept – they do little to securely prepare the ground for human trials and may give misleading data about a drug’s safety and efficacy.
With respect to product development, early priorities include the need to achieve viable drug stability and bioavailability by:
- Identifying the most appropriate dosage form – powder or liquid
- Assessing the effect of alternative excipients – on stability, retention at the deposition site and/or permeation
- Determining the impact of the deposition profile and the ability of different devices to achieve the desired delivery
- Investigating the effect of patient technique.
Liquid-based formulations tend to be easier to develop, for both intranasal and pulmonary delivery, but the merits of dry powder formulations can be compelling: higher drug loading, greater stability and increased retention at the deposition site. For certain drugs, such as those that are poorly soluble or labile biologics, these benefits may be pivotal to the realisation of a commercially viable product.
Solubility or stability enhancers may facilitate a liquid formulation, but there is a distinct lack of approved candidates for these delivery routes. The lack of permeation and retention enhancers (mucoadhesives) is similarly problematic for all dosage forms. While the use of novel excipients is an option, this will add time and risk to the product development process. Currently, there is no requirement for OINDP formulations to be sterile, but the need to maintain microbial limits in line with regulatory guidelines routinely necessitates the inclusion of preservatives, which may negatively impact stability, adding to the complexity of the formulation process.
At the preclinical stage, stability requirements only extend to ensuring the consistency of the product for the duration of clinical trials, which could potentially be just a few months. However, given that a poorly stable product will inevitably need to be reworked, should clinical trials prove successful, it is worth the investment to achieve robust stability early, so as to minimise risk and timelines.
On the other hand, when it comes to the regulatory tests associated with OINDP characterisation, there is scope for discernment versus the full CMC guidance from the regulators, most notably during Phase I clinical studies. There is a balance to be struck between gathering enough data to help make decisions or justify changes down the line and doing everything too soon, then having to repeat it again later. Figure 2 differentiates critical tests from those that are nice to have, helping to focus efforts to achieve the maximum effect. Time and cost can also be saved by adopting a strategy of “phase-appropriate method qualification”, where there is no need to fully validate analytical methods at this stage, though they must, of course, be robustly fit for purpose.
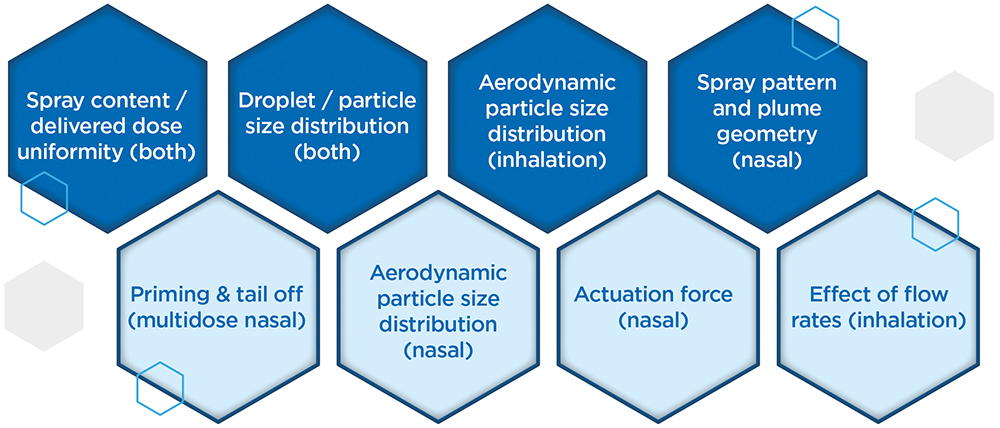
Figure 2: Top line (dark blue) OINDP characterisation tests are critical while those on the lower line (light blue) may be delayed in preclinical or early clinical studies to minimise the risk of duplication associated with product refinement.
CLINICAL: BUILDING THE CASE FOR REGULATORY SUBMISSION
A primary focus going into clinical trials is setting a schedule for gathering the remaining information required for regulatory submission. Unfortunately, for OINDPs, such efforts are hampered by a lack of harmonised and, in some instances, up-to-date guidance. This situation is at its worst for nasal powders, where there is only minimal specific information available.
Phase I
Planning out how much drug product and how many units will be needed to implement clinical trials, and how they will be made, is a crucial task for early-stage clinical studies. Even though the clinical study is small, it is easy to underestimate how many units are needed for the method qualifications, release and stability studies.
For OINDPs, the manufacture of drug substance and drug product are often discrete processes with no stated requirement for sterility in the assembled OINDP. Manual filling and assembly at the clinical trial site is therefore an option for Phase I, provided that there is sufficient expertise to do so. This approach can be advantageous if, for example, stability is poor or not yet known, or if drug cost or scarcity prohibits early commercial-scale processing.
Ensuring that a suitable reference standard is in place for the drug substance as early as possible is also helpful, particularly for new molecular entities. Without this, there is a risk of delays in establishing effective analytical protocols, as well as the potential to undermine confidence in the ongoing stability of that product, particularly as the drug substance and drug product manufacturing processes scale up and advance through the clinical stages.
“In the early stages, when there are few units to test and the product is subject to change, it can be advantageous to designate data for characteristics unassociated with product safety, such as nasal spray pattern, as “for information only” and delay specification setting.”
Phase II
Phase II marks the point at which commercially representative processes become essential and the demand for drug product begins to grow quickly, with larger scale stability studies getting underway alongside more extensive product characterisation. Extractables and leachables studies should be on the radar at this point, as the material for such studies is most efficiently produced as part of the registration batches. Data relating to extractables should be available from component suppliers, but leachables studies are product-specific and may be required even for generics.
Now is also the time to investigate in detail how patients will use the device, as regulators will expect to see evidence that human factors have been considered in the device choice and in the development of effective instructions for use. The goal is to demonstrate that patients can use the device as instructed and, if they do, that it delivers well-defined performance.
Phase III
Moving into Phase III, manufacturing ramps up considerably. Representative registration batches are typically produced at 1/10th commercial scale, with additional batches produced in association with pilot-scale studies, scale-up and for process validation. Batch release testing requirements rise correspondingly, and full-method validation becomes necessary in accordance with USP/ICH guidelines.
A stability programme to establish shelf life is an important aspect of Phase III trials that calls for the testing of three batches (even for ANDAs) under room temperature, accelerated and intermediate storage conditions. Furthermore, there is a need to assess the impact of device orientation during storage, which doubles the amount of product required for testing.
Lastly, this is the point at which one off product characterisation studies for OINDPs are necessarily implemented. For nasal sprays, for example, such tests include robustness (drop and vibration testing/cleaning), temperature cycling, priming/repriming, photostability, effect of dosing orientation and tail-off or profiling (Figure 3).
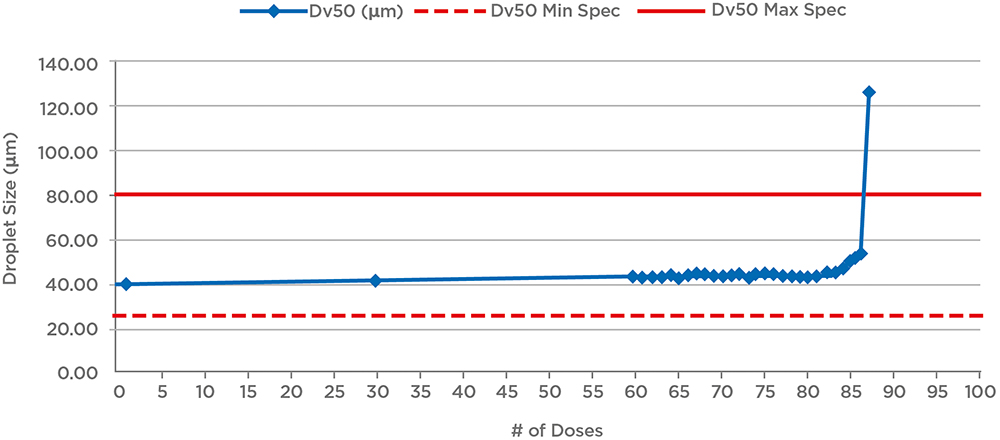
Figure 3: Example tail-off data for a nasal spray shows that performance is maintained for an additional 25 doses beyond label claim (60 doses). Reducing fill levels for the device would therefore reduce waste and the risk of confusion for patients; a relatively short tail-off is preferable.
CLINICAL: CONSIDERATIONS AND CHALLENGES
One issue to consider in clinical trials is when to establish specifications for the product, and for which characteristics. In the early stages, when there are few units to test and the product is subject to change, it can be advantageous to designate data for characteristics unassociated with product safety, such as nasal spray pattern, as “for information only” and delay specification setting.
Regarding product changes, the likely commercial device should ideally be in use for Phase II – preferably in Phase I, as well – but device changes are sometimes unavoidable and can be managed, however, having solid characterisation data to support the bridging will help a lot. For example, Phase I trials may indicate a need for dose escalation, necessitating a switch from a single-dose to a multidose device, or amplify the benefits of powder formulation. Unfortunately, any change in the device may change in vivo deposition behaviour and, by extension, may impact clinical efficacy, so this will need to be evaluated. Robust characterisation of whatever device and formulation is taken into the clinic provides the best possible foundation for justifying any such change, should it prove necessary, and can therefore be a worthwhile upfront investment.
The number of product units required to implement OINDP clinical trials is considerable, and routinely underestimated. The schedule shown in Table 1 indicates the number of products needed for just one test – aerodynamic particle size distribution (PSD) – for a single nasal spray batch, for a three-year stability programme; 25°C/60% relative humidity (RH) is the room temperature condition, 40°C/75%RH is the accelerated condition, 30°C/65%RH is the intermediate condition and 30°C/75%RH is an additional test condition associated with the ambition of marketing the device in tropical climates (ICH Zone 4).
| Condition | Orientation | 0 | 3M | 6M | 12M | 24M | 36M | Extras | Number of Units |
| 25°C/60%RH | Upright | 10 | 10 | 10 | 10 | 10 | 10 | 10 | 60 |
| 25°C/60%RH | Inverted | 10 | 10 | 10 | 10 | 10 | 10 | 60 | |
| 30°C/75%RH | Upright | 10 | 10 | 10 | 10 | 10 | 10 | 60 | |
| 30°C/75%RH | Inverted | 10 | 10 | 10 | 10 | 10 | 10 | 60 | |
| 30°C/65%RH | Upright | 10 | 10 | 10 | 10 | 10 | 10 | 60 | |
| 30°C/65%RH | Inverted | 10 | 10 | 10 | 10 | 10 | 10 | 60 | |
| 40°C/75%RH | Upright | 10 | 10 | – | – | – | 4 | 24 | |
| 40°C/75%RH | Inverted | 10 | 10 | – | – | – | 4 | 24 | |
| Total Number of Units | 10 | 80 | 80 | 60 | 60 | 60 | 68 | 408 | |
Table 1: An example for the number of units required solely for DSD measurements associated with a stability test for a nasal spray illustrates how the number of units required for clinical trials builds up.
Aptar Pharma’s experience indicates that it is realistic to expect to need at least 15,000 units for multidose devices for Phase III trials, more if using unit dose, with material-hungry tests, such as microbiological testing and the assessment of leachables, adding to this requirement. It is key to plan out these requirements sufficiently early to secure registration batches of the required size and establish efficient routes for production, as any failure to do so can have a major impact on progress to market.
OINDPs are arguably the most challenging class of pharmaceutical product to make, and relatively few contract design and manufacturing organisations are equipped to support the transition from R&D into a GMP environment. Be sure to determine whether a potential partner rigorously understands specific drug sensitivities, the intricacies of the device and the analytical demands associated with the product before committing to avoid compromising clinical trials.
To learn more about Nanopharm’s specialist respiratory offering, visit: nanopharm.co.uk.
CONCLUSION
The amount of product required for OINDP clinical trials may well surprise developers more familiar with alternative dosage forms, as indeed might the many and diverse issues associated with bringing OINDPs to market. The complex interplay between device, formulation and patient that drives drug delivery via the nose and lungs presents a major challenge; understanding how to manipulate device and formulation characteristics to propel a promising drug candidate to success remains an expert task. There is significant and exciting clinical potential in this field, and growing interest in exploiting it. Experienced, specialist, integrated CROs and contract design and manufacturing organisations can help product developers to invest well, at the right time and in the most productive way to reduce the risk associated with these routes and accelerate successful products to market.
