
To Issue 129
Citation: Gonzalez M, “From Vial to Prefilled Syringe: Migrating a Drug Product Presentation”. ONdrugDelivery Issue 129 (Feb 2022), pp 31–33.
Martin Gonzalez discusses some of the most relevant engineering activities that are necessary to transfer a product successfully from a vial into a prefilled syringe presentation.
There is a growing trend for pharmaceutical companies to ask their CDMOs to support the conversion of an existing product presentation into a different final container type – more specifically, to provide options for moving a drug product from a vial presentation into a prefilled syringe (PFS).
“The decision is increasingly being driven by a desire to create more convenient drug products, easing or tailoring administration to the therapeutic need.”
WHY TRANSITION FROM A VIAL TO A PFS?
There are many reasons why a drug developer may be interested in converting a drug product currently provided in a vial form into a PFS. It is sometimes driven by quality requirements to preserve product safety or potency. However, more often than not, the desire to create more patient-centric administration formats and to extend patents is underpinning the move.

Figure 1: Prefilled syringes remain one of the fastest-growing classes of drug delivery device.
In addition, the decision is increasingly being driven by a desire to create more convenient drug products, easing or tailoring administration to the therapeutic need. When an injectable’s intended use is during emergency surgery, in doctors’ surgeries or in a variety of at-home care scenarios, simple and/or more rapid administration is often required. Ease of use can positively impact patient compliance, which is one of the longest standing problems in healthcare. PFS (Figure 1) remain one of the fastest-growing classes of drug delivery device, due to advances in technology and increased development of parenteral drugs.1 The core rationale for switching from a vial to a PFS is that it helps businesses manage the brand lifecycle of their products, offers significant benefits to healthcare providers (HCPs) and patients, and can help cut manufacturing and product costs. Manufacturers increasingly need to innovate to retain market presence or strengthen their market share for an indication.
On the manufacturing front, vials are sometimes overfilled to ensure the full dose is retrievable for administration to the patient. This is not necessary with PFS, as they inherently improve dosing control, significantly reducing drug product waste. As a result, PFS have lower manufacturing costs overall.
COMPARING GLASS VIAL AND PFS PROCESSING

Figure 2: A vial fill line usually uses in-house processed glass before the vial enters the aseptic fill suite.
Glass Vials
Commodity Preparation
Vial and PFS fill lines are inherently different. A vial fill line (Figure 2) usually uses in-house processed glass before the vial enters the aseptic fill suite hosting the filler inside a restricted access barrier system (RABS). The glass vials are received at the facility from the glass manufacturer “as is”. Once received, inspected and released, the vials are transferred to a washing-depyrogenation continuous operation tunnel. There, vials are washed with chilled water for injection (WFI), drained and dried out with filtered air or nitrogen gas before entering a depyrogenation oven where they are dry heated at temperatures in the range of 275–350°C. They then move to cooling stations under filtered air or nitrogen gas to reduce their temperature to near room temperature. Depending on the vial size, the washing-depyrogenation line speeds can vary to accommodate the fill line output and to obtain a correct depyrogenation process.
Common issues noted with this process include:
- Introduction of particulate matter
- Increased glass delamination, usually detected after product has been placed on stability for months or years
- Incorrect washing-depyrogenation tunnel speed set-up leading to surface scratches resulting in high numbers of rejected units.
Vial Feeding into Fill Suite
Once vials are washed-depyrogenated, they move to a Class II/Grade B classified area accumulation table. From there, vials enter the RABS to be filled (Class I/Grade A area) and capped, then moved to visual inspection stations or into lyophilisers through HEPA carts or an automated loading-unloading systems (ALUS). The fill process is performed under air or nitrogen atmosphere, depending on the drug product needs. Vapour hydrogen peroxide (VHP) is used to sanitise the line, followed by a period of venting/aeration to remove potential VHP traces. In some cases, a manual decontamination process is used, and steam-sterilised change parts are fitted prior to the introduction of vials into the RABS. For liquid products, the vials are fully capped and crimped, then moved to visual inspection stations for a manual, semi-automated or automated visual inspection process. If the product is lyophilised, the vials are partially capped, placed in carts (manually or into ALUS carts) and loaded into lyophiliser chambers. Visual inspection occurs after the product has been lyophilised, capped and crimped.
PFS
Commodity Preparation
PFS come as a sterilised, pre-processed, nested commodity. PFS manufacture has improved tremendously over the last couple of decades and now provides a very consistently high-quality product. Rejection limits due to cosmetic factors have been significantly lowered, mostly because each PFS glass barrel is isolated from its neighbours, which are all inserted in a template inside a plastic tub.
The manufacturer’s processing of PFS involves the washing, siliconisation and sterilisation processes using gamma irradiation. In addition, the tubs are single or double bagged with plastic wraps. Once the PFS arrive at the CDMO manufacturing site, they are typically released for manufacturing, usually by undergoing a few more tests than a vial would undergo. Units are tested for sterility, correct needle attachment, unclogged needle and tip cap removal force, on top of the testing common to vials (dimensions, type of glass determination, hydrolytic resistance and cosmetic defects). There is no further processing for the PFS before heading to the fill line at this point, as opposed to the washing-depyrogenation required for glass vials.
PFS Feeding into Fill Suite
Feeding the commodity into the fill suite is where the process differs the most between glass vials and PFS. To insert the nested PFS (tubs) into the fill line, the tub – which usually comes double bagged – must be disinfected first.
In a grade B/C area, the outer bag is removed, then it enters the RABS filler system through a conveyor belt where the inner bag is removed under a class I/grade A area, and then it goes into the electron-beam (E-beam) tunnel. A conveyor belt takes the tub through a path inside the E-beam tunnel where low-energy electrons bombard the surface, killing any biological entity present, and then the tub’s Tyvek® lid is removed. The removal of the outer bag can still allow micro-organisms to be carried into the aseptic area, and the purpose of the low-energy E-beam treatment of the tubs is to inactivate any such contaminating micro-organisms. A simple schematic of an E-beam/RABS fill-finish system is shown in Figure 3.
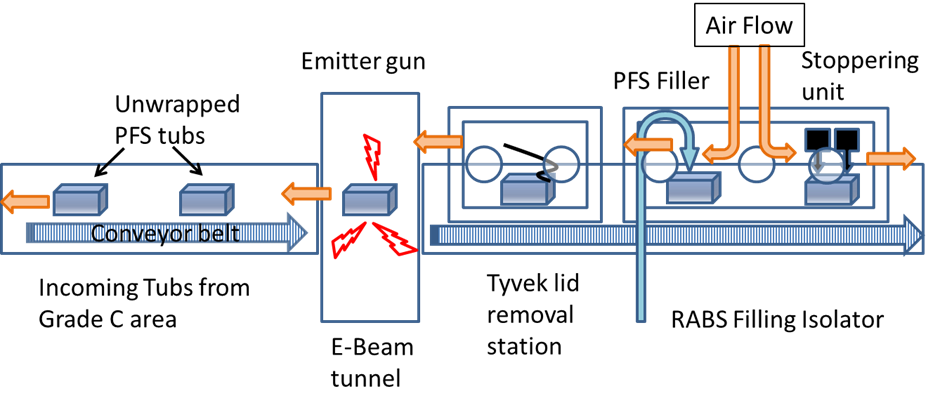
Figure 3: A schematic of an E-beam/RABS fill-finish system.
In manual operation modes (where operators remove the outer bag and place the tub on the conveyor), bioburden numbers of around 100 colony forming units (CFUs) per tub are common (Bachmann and Harper, 2007). But in automated operations a dramatic reduction of CFU count is usually found. The radiation dosage is established, considering the material comprising the outer surfaces of the tub, as well as any effects on the PFS constituent materials inside the tub. The physical and chemical properties of polymers are affected by irradiation, and the radiation dosage should be selected so that its effects are minimised while ensuring proper sterilisation.
MOVING DRUG PRODUCT FROM VIAL INTO PFS
“To transition a drug product from a vial into a PFS, developers must first consider the physicochemical properties of the drug product and the impact on its quality as it is in contact with the different components of the container closure. When evaluating a new container closure.”
Analytical Requirements
To transition a drug product from a vial into a PFS, developers must first consider the physicochemical properties of the drug product and the impact on its quality as it is in contact with the different components of the container closure. When evaluating a new container closure, in this case a PFS system, screening studies for suitable parts are needed. There are many factors involved during the selection process:
- Nature of the drug product (small molecule or biologic)
- Formulation composition (high salt concentration and high solution pH prompts glass delamination)
- Sensitivity to oxygen in the headspace or oxidants, like tungsten traces present in some PFS types (can affect a biologics’ potency and purity)
- Headspace volume (affecting plunger stopper movement during shipping, thus compromising closure integrity)
- Siliconisation levels (affecting device functionality or promoting aggregate formation on sensitive biologics)
- Container closure integrity testing using suitable methods. High voltage leak detection (HVLD) might sometimes not be suitable for biologics, but fine for small molecules. Headspace analysis by frequency modulated infrared spectroscopy (e.g. Lighthouse) are both high-throughput methods. Dye ingress or pressure decay could be easily implemented for most container formats but are low-throughput and destructive methods that can only test a few units, rather than an entire lot.
Availability of analytical test methods is important to carry out suitability studies of the different commodities (glass barrel, plunger stopper type, etc). Ancillary facilities and equipment (stability chambers, compounding capabilities for lab batch scales, specific analytical instrumentation) are also necessary, otherwise delays could happen when transferring samples to a third-party testing facility or the pharmaceutical company’s own laboratories.
“Establishing as early as possible what experimental materials are available and the associated timescales will ensure a smoother rollout.”
Engineering and Commodities
Screening activities are carried out in process development laboratories, while engineering/machinability trials are required to test the selected parts in the specific fill line with the exact commodities. The screening activities to select product-contact parts are, under normal circumstances, completed within 4–6 months. It is then when the programme can proceed to machinability or commodity suitability trials in the fill line. When starting these types of screening studies, it is vital to check commodity availability with the vendor. Establishing as early as possible what experimental materials are available and the associated timescales will ensure a smoother rollout. It also helps define the extent of the design of experiments space to carry out.
A manufacturing subject matter expert team provides the necessary knowledge to select the right parts that will be needed to process the commodities and will lead the activities to completion.
Some of the critical activities to perform are dependent on the current fill line configuration:
- If nitrogen blanketing is needed, a full vacuum stoppering retrofit or a vacuum-assisted stoppering (VAS) process could be installed. Some coated plungers might be prone to wrinkling when inserted into the barrel, so additional trials might be needed to set up the fill line correctly. These activities can be completed in around 5–6 months after receiving the necessary parts from the filler’s manufacturer. Scoping out the project needs might take some additional time – up to three months in some cases.
- If a change in PFS size is needed (e.g. if the line is set to 1–2.25 mL), then going to a different (e.g. 5 mL) syringe size will require change parts to run them. It’s important to consider having multiple sets to facilitate quick line turnaround for line set-up and as safety back-ups.
- Fabricating and delivering the vibrating plunger sorting bowl takes around 25–28 weeks. This requires the plunger stopper to be identified before ordering the fabrication.
- Vendor installation of required change parts, developing the filler format recipe, installing and fitting change parts, and ensuring solution volume accuracy and plunger placement requirements often takes one or two weeks to complete.
- If the PFS fill line is equipped with E-beam capability (most are) and tub size change is required (e.g. going from a 10 x 10 PFS distribution format to an 8 x 8 type, or from a 4-inch-high to a 6-inch-high tub size), the controlling software and the handling parameters will need to be programmed to handle the new tub size/format.
- Trial testing of the sorting bowl requires approximately 50,000 plungers. For the change parts trials, another 70,000 PFS/plungers/rods sets are needed as well.
- Automated (or manual) inspection activities also need to be considered at this time. Developing an automated container inspection process will require thousands of samples to carry out feasibility and recipe development studies. These activities normally take a few months (three to four) to complete.
All new line parts will require new validation activities, so it is best to involve validation teams as early as possible to ensure a correct and appropriate approach is applied. Additionally, project timelines can be impacted by commodities supply lead times. To initiate engineering/machinability trials, a sizeable quantity of PFS, plungers and rods are needed. A lead time of 8–10 months for parts is not unheard of. Engaging with the vendor at an early point might ensure the timely supply of the required quantities.
“Adopting a new final container takes concerted efforts from product development, incoming quality, chemistry quality, engineering, manufacturing, validation, procurement and regulatory teams.”
BRINGING AN IMPROVED PRODUCT TO MARKET TAKES A VILLAGE
Adopting a new final container takes concerted efforts from product development, incoming quality, chemistry quality, engineering, manufacturing, validation, procurement and regulatory teams. Seamless co-ordination within the CDMO and the pharmaceutical company is paramount, since the whole process – all the way up to adopting a new container closure (or conversion to another) – could easily take up to two years. This is just to the point of manufacturing clinical trial material/registration batches to place on formal stability programmes to ensure the selected container is suitable for the shelf life of the drug product.
REFERENCE
- “Global Prefilled Syringes Market Size: Growth, Trends and Forecast.” Press Release, ResearchandMarkets.com, April 23, 2020.