Citation: Miles B, “Respiratory Drug Delivery: Then, Now and Soon”. ONdrugDelivery Magazine, Issue 106 (Apr 2020), pp 6-9.
Brennan Miles takes a look back at respiratory drug delivery over the last few decades to see what we can learn from the past – and looks ahead to what the future might look like, covering sustainability, device regulation, and formulation milestones.
Respiratory drug delivery has come a long way in the last few decades, with a raft of changes in legislation, standards and industry trends. Whilst it’s impossible to predict the future, it’s interesting to take a look back, explore how our industry has progressed over the last 30-plus years and consider what areas we are likely focus on in the coming decades.
ENVIRONMENTAL CONCERN AND SUSTAINABILITY
“Should we potentially sacrifice patients’ disease management by withdrawing or changing devices that are environmentally damaging? Or should patient health take priority over environmental protection?”
Environmental and sustainability concerns are hot topics for the respiratory drug delivery sector – and have been, in one form or another, for some time. Environmental factors such as air quality can have a huge impact on respiratory conditions, and improvements in the treatment of existing diseases, so it’s a hugely important area for the industry in general. A drive towards protecting the environment in the late 1980s triggered one of the biggest recent changes in the respiratory industry and – whilst the conversation around the environmental impact of the industry has since moved to encompass other areas – it remains a huge driver for change.
In 1987, the Montreal Protocol was agreed and signed by 167 countries, due to growing global concerns over the depleting ozone layer. The agreement was designed to protect the environment by phasing out the use of harmful substances, including chlorofluorocarbon (CFC) propellants which were widely used in pressurised inhalers at the time. This change had two important effects: it triggered development of alternative hydrofluorocarbon (HFA) based propellants and prompted more research effort into other device-reliant drug-delivery methods such as dry powder inhalers – which work without propellants, instead using patients’ respiratory effort to aerosolise the drug.
The effect of the shift away from CFCs is clearly visible in contemporary devices, and there has recently been much discussion in the industry around HFA alternatives to further address sustainability and wider environmental concerns. Some types of HFAs, whilst not as damaging to the ozone layer as CFCs, still represent a potent greenhouse gas, and calls for discontinuing or at least limiting their use are growing. The problem with this, however, is that whilst current propellant-based inhalers may be detrimental to the environment, they are also an important part of the lives of hundreds of thousands of patients worldwide.
This raises a big question – should we potentially sacrifice patients’ disease management by withdrawing or changing devices that are environmentally damaging? Or should patient health take priority over environmental protection? There is no easy answer but significant effort is being spent to find a reasonable solution, including the development and approval of more environmentally friendly HFA propellants and other inhalation technologies.
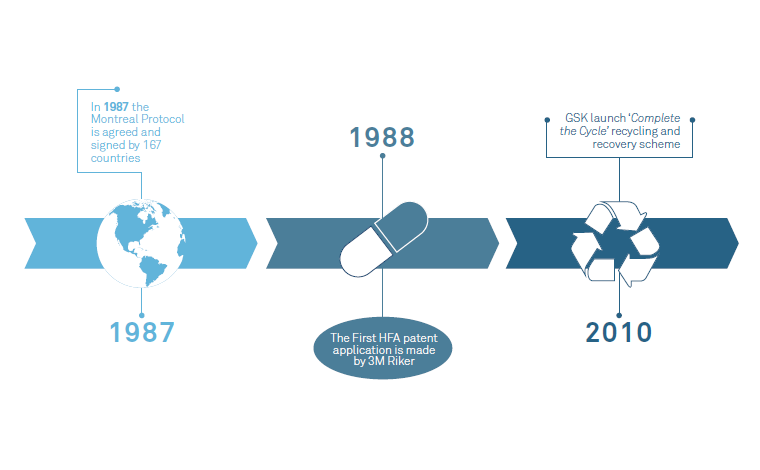
Consumer product companies have been making serious moves for several years to improve the environmental impact of products and packaging. These include switching to recycled or sustainable materials and reducing the use of single-use plastics. The medical industry necessarily tends to move slower than our consumer counterparts – generally about 10 years behind consumer sector trends – but sustainability has been firmly on the respiratory agenda for a few years now (Figure 1). It is clearly a growing driver, with companies and countries alike making big commitments to sustainability initiatives.

Figure 1: Environmental milestones in respiratory drug delivery.
In 1987, our main concern was the damaged ozone layer. But today there are many other concerns that affect the environment, such as the prevalence of plastics, particularly in single-use devices. In medical device development it’s easy to overlook the positive environmental impact that small changes can make, as patient safety comes above all other issues. However, devices like inhalers that are produced in very high volume provide great opportunities for reducing environmental impact in low-risk areas. Some examples include developing smarter packaging concepts to reduce bulk and weight as well as making use of the wide range of sustainable materials that are already available. We can expect to see more focused effort in the early design phases to identify low-risk areas where alternative – or perhaps recycled – materials can be used without presenting a risk to the patient.
More thought is also being given to how devices are managed at the end of their lives, recovering devices to recycle the materials used (rather than incinerating them as usually happens). Inhaler recycling schemes are being introduced in many areas – Teva recently launched one such scheme in Ireland, and GSK has recycled 1.2 million inhalers as part of its Complete the Cycle recycling and recovery scheme since 2010.
Typically, major shifts in the medical devices industry only come about when pushed by regulation, and we should expect some developments in this area soon. There remains the complex underlying issue of finding a balance between patient health and environmental health, especially when each affects the other.
REGULATION, REGULATION, REGULATION
Many of the standards and regulations that we take for granted in the medical devices industry today have existed in their current form for less than 30 years. The Medical Devices Directive (MDD 93/42/EEC) was first adopted in 1993 to harmonise laws relating to medical devices within the EU. Prior to the MDD, every European country had its own laws, regulations and different ways of approving medical devices, although mutual recognition agreements were an effective way to allow devices approved in one territory to be approved across many. Operating under the same regulations across multiple European countries is beneficial to pan-European corporations because it streamlines the regulations device developers need to adhere to.
The MDD has undergone several amendments over the years (see Figure 2) but the largest change will happen shortly when it transitions into the Medical Devices Regulation (MDR). There are some significant changes in the new regulation, such as a shift to a more complete lifecycle approach, and an increased number of safety requirements: the word “safety” appears 290 times in the MDR compared with only 40 in the MDD.

Figure 2: Key medical device regulations introduced since 1993.
Another key regulatory milestone in the industry came in 1996 when ISO13485 was published – a standard that forms the basis of quality management systems for European medical device developers and manufacturers. The MDD talked about the need to have a quality management system in place but ISO 13485 describes what that means in practice and plays a hugely important role for medical device design, development and manufacturing. Before this, manufacturers typically deferred to the principles of ISO 9001, which established the basic requirements for a supplier to assure product quality but was not specific to the medical devices industry.
“Whilst there’s a lot to be learned from landmark successes in respiratory drug delivery such as TOBI, there’s perhaps even more to be learned from failures.”
The year 1998 saw the introduction of ISO 14971, covering the requirements for risk management during medical device development and post-production. It has been an immensely important standard for defining risk for medical devices – the combination of probability of occurrence of harm and the severity of that harm – so that risks can be identified and mitigated in a systematic way. Patients do and should take for granted that medical devices are safe for them to use and this is exactly why good risk management is so important for medical device developers.
Specifically, for inhaler devices it was only in 2003 that the US FDA issued its guidance for “integration of dose counting mechanisms into MDI products”, which prompted an abundance of new dose counter innovations for inhalers. In 2009, ISO 20072 was published which describes device design verification requirements and test methods. The standard requires some specific and challenging environmental conditions to be maintained for the testing procedures. It will be interesting to see what innovations new and future standards prompt in our industry.
FORMULATION MILESTONES
There is no typical length of time that it takes for a new drug to be tested and approved. The best rule of thumb is that it can take anywhere from 10 to 15 years for an experimental drug to move from preclinical testing in the laboratory to gaining regulatory approval and use. It has been estimated that only one in every 5,000 new drugs makes it to market. With odds this bad, it is remarkable that so many of the most valuable respiratory formulations (Figure 3) were discovered in recent years.

Figure 3: Respiratory drug formulation milestones.
In 1990, Glaxo (now GSK) launched Serevent (salmeterol) – the first long-acting beta-agonist (LABA) for the maintenance of asthma and COPD. This was followed in 1998 by Seritide (salmeterol + fluticasone propionate), which is marketed as Advair in the US. Seretide/Advair remains the best-selling asthma treatment of all time, generating revenues of over £5.7 billion.
You can’t discuss inhalation formulation advances without mentioning Sir David Jack (1924–2011). He was the exceptional research scientist behind many of the blockbuster respiratory medicines that came out of Glaxo over the years, including Ventolin (salbutamol), Serevent, and Becotide (beclomethasone dipropionate). These drugs are still widely prescribed to treat asthma and, even decades later, no better equivalent exists. Sir David even continued to carry out research work after his retirement from Glaxo. As recently as 2011 the drug development company Verona Pharma announced that it was seeking commercial licensing agreements for Ensifentrine, an anti-asthma and hay fever treatment developed by Sir David as an alternative to conventional steroids and beta-agonists.
“Medical device companies and pharmaceutical firms have come to understand that the trade-off between sustainability and profit is an outdated concept.”
Another milestone in respiratory drugs that deserves a mention is tobramycin solution for inhalation (TOBI) which was approved in 1997 for the treatment of cystic fibrosis (CF), which severely affects the function of the lungs. TOBI was the first inhaled antibiotic for the treatment of CF and has been credited with significantly extending the life expectancy of CF patients. TOBI represents one of the few success stories in systemic inhaled therapies, outside of asthma and COPD treatment.
Whilst there’s a lot to be learned from landmark successes in respiratory drug delivery such as TOBI, there’s perhaps even more to be learned from failures. Exubera – the first inhaled insulin to be approved by the FDA – is in the latter category of respiratory milestones. It represents one of the most interesting examples in recent history of how a poor understanding of patients’ needs and a difficult-to-use device ultimately led to a major failure to gain market acceptance. Following a 2006 launch by Pfizer, it was eventually withdrawn at an estimated loss to Pfizer of $2.8 billion (£2.3 billion). It’s difficult to pinpoint the reasons Exubera wasn’t successful but a combination of fundamental issues meant that it didn’t follow in the footsteps of TOBI as a successful inhaled systemic therapy.
PREDICTING THE FUTURE
There has been so much innovation with respiratory medicines and inhaler devices over the last 30 years that it is impossible to guess where the industry is headed in the next 30 years. We can, at least, think about what is likely to happen in the foreseeable future and how some of the current hot topics such as sustainability, regulation and formulation innovation may evolve.
In the past, many businesses have viewed their commitment to improving the environment as something that should be addressed when they are instructed to, rather than seeing it as core to their values and strategy. This is now changing. Medical device companies and pharmaceutical firms have come to understand that the trade-off between sustainability and profit is an outdated concept. They are now reacting to the world around them and recognise that environmental concerns are also their (and their shareholders’) concerns.
Good opportunities exist for making medical devices greener: focusing on the manufacturing process to reduce production waste and reduce the need to ship materials, components and sub-assemblies to different manufacturing sites; specifying recycled (or recyclable) materials during the development of new devices; and reducing the device packaging that is delivered to the patient (for example, by combining packaging and instructions for use and by using simple cardboard support trays to replace plastics).
Another approach is to reduce the complexity and physical mass of the devices. A few years ago, Team Consulting put together a concept for a novel inhaler that was essentially a cardboard tube with a simple piercing mechanism to release the drug from an innovative single-dose blister. Pioneering solutions like this should be seriously considered in the next few years to supplement current respiratory device offerings. We need to encourage more focus on sustainable thinking in the early phases of a new product design in combination with programmes that deal with the collection and recycling of used devices. Expect to see some real changes in this area soon.
Improving patient adherence is clearly a major theme for the delivery of inhaled medicines. Worldwide, non-compliance is a major challenge to the delivery of healthcare. Adherence is also closely linked to sustainability because the better patients follow their medical instructions, the less wastage and overuse of medicines and devices there are. The industry is attempting to tackle this through new technologies, such as companion apps that give patients reminders to take their drugs, and device training instructions. These can also work alongside the emergence of smart devices that actively assist the patient to inhale correctly to improve the drug delivery effectiveness. New technology also has a downside because it increases device complexity, so the benefits need to be carefully weighed and balanced to decide how and where the technology is used to be most effective.
It’s incredibly difficult to predict where the new inhaled therapies are going to come from and what they are going to be. We do know that inhalation of drugs into the lungs is one of the least invasive routes and works well for topical therapies to treat respiratory disorders. It is perhaps somewhat astonishing that more systemic therapies have not been developed for the inhaled route, possibly in part because of the failure of Pfizer’s Exubera programme. Reassuringly, there have been some recent developments in this field – in 2014, for example, the FDA approved a new inhaled insulin (Afrezza) to treat diabetes.
We also know that good clinical outcomes in respiratory medicine rely on a combination of drug formulation and delivery device that is simple to use and produces good deposition of the drug inside the lung. Recent new device technology development activities have seen an emergence of improved nebulisation devices and a renewed interest in liquid mist inhalers. Technologies such as this have the capability to deliver more complex formulations which would have otherwise been impossible in a dry powder form. These new device technologies may well be the key that opens the door to new inhaled therapies beyond asthma and COPD.