






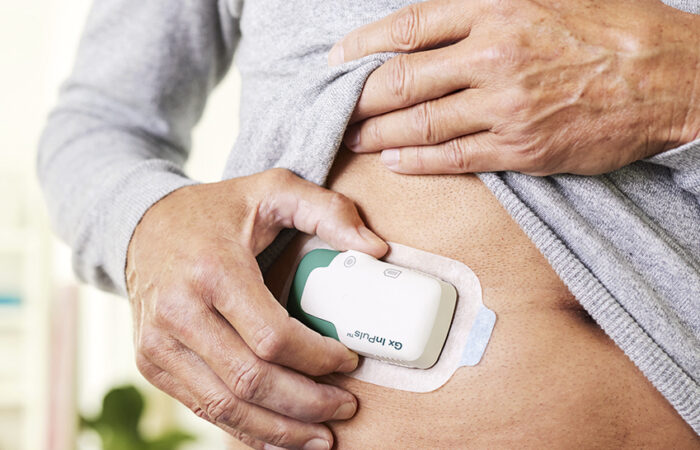





















See what ondrugdelivery's readers and contributors think

I‘m working on the development of microparticles for the release of veterinary antibiotics, and polymeric nanoparticles for siRNA and chemotherapeutic delivery. ONdrugDelivery has been a very usefull platform to connect the scientific research, and marketed drug development.
Your website and publication is a great asset for me. I’d recommend your publication to my colleagues. I am sure that we will gain knowledge and update information through your timely report. Thanks for making it available.
ONdrugDelivery Magazine is a great resource that brings a lot of value to the work my team and I are doing in the area of drug delivery. The device-specific content as well as the broader discussions surrounding industry trends and issues are extremely informative and very well written.
I picked up your latest copy at the Boston PODD event and read it with great interest. You’ve really come a long way and you put out an amazingly professional and value-packed magazine.