
To Issue 182
Citation: Eden O, Webb T, “Developmental Advantages in Injectable Combination Drug Products”. ONdrugDelivery, Issue 182 (Jan 2026), pp 114–117.
Visit Jabil at Pharmapack Paris! – Stand 4D101
Oliver Eden and Travis Webb consider how regulatory considerations and patient-centric device designs are transforming the landscape of self-administered injectable drugs.
One of the biggest changes in pharmaceuticals over the past 10 years has been the increase in self-administered injectable drugs such as breakthrough medicines for chronic conditions. This transformation goes beyond process change and represents a strategic advantage for developers that embrace early collaboration between drug formulation scientists and device engineers.
“THE DAYS OF DEVELOPING DRUGS AND DEVICES IN SILOS ARE OVER. INSTEAD, PHARMA BRANDS MUST FOSTER ONGOING DIALOGUE BETWEEN FORMULATION SCIENTISTS, DEVICE ENGINEERS AND REGULATORY EXPERTS FROM THE OUTSET.”
The intersection of formulation science, device design, regulatory considerations and patient-centric clinical trials is now the crucible in which successful therapies are forged. The days of developing drugs and devices in silos are over. Instead, pharma brands must foster ongoing dialogue between formulation scientists, device engineers and regulatory experts from the outset.
THE RISE OF SELF-ADMINISTRATION
An increasing prevalence of chronic diseases, patient preference for convenience and advances in delivery technologies, such as autoinjectors, are transforming how therapies are administered. As biologics and other novel formulations become more common, treatment is shifting beyond traditional healthcare settings to the hands of patients themselves. Every pharma company would likely offer medicines in a simple pill if they could; however, many biologics cannot be formulated as oral tablets because they break down or lose potency in the gastrointestinal tract.
As a result, developers are advancing injectable and device-based delivery options that ensure therapeutic effectiveness. In a real sense, the drug determines the device. As patients seek therapies that align with their daily routines, pharmaceutical companies are responding with prefilled syringes (PFSs), autoinjectors and wearable devices that simplify administration while supporting patient retention, adherence, compliance and persistence.
Self-administration is now a key focus, with autoinjectors and on-body devices designed to make dosing intuitive and convenient. Devices must accommodate patients with needle-phobia or limited dexterity, along with maintaining a consistent dosing regimen. Pharma companies are increasingly aware that device usability directly impacts adherence. If a patient struggles with a device or fails to complete an injection, therapeutic outcomes worsen.
The mission with self-administration is not just to persuade patients to inject themselves but also to ensure that, when they do, they follow the provider’s instructions. You can provide a patient with an injection device that they are happy to use, only to find that they lift the device from the skin halfway through the injection sequence and only deliver half the dose. The goal is to produce products that empower patients to take control of their own dosing with devices that are not just easy to use, but are easy to use correctly.
The rise in self-injections has also affected regulatory agencies such as the US FDA and EMA, which have responded by intensifying their focus on the patient experience and usability. Recent FDA guidance has emphasised the need to consider human factors early in development to ensure that combination drug products are intuitive, safe and effective for patients. Common themes across regulatory bodies for combination products include early classification, integrated risk management for both drug and device, human factors and usability studies, and labelling and instructions.
FORMULATION AND REAL-WORLD FUNCTION
Early integration of formulation and device development creates products that are stable, user-friendly and compatible with delivery systems optimised for real-world use. There are also formulation aspects that help with patient convenience. For example, long-acting injectables allow patients to reduce the required number of doses. This cross-disciplinary approach is essential for pharma companies to meet regulatory expectations, enhance patient satisfaction, and ensure speed and success in a competitive landscape.
A combination product, as the name suggests, combines three different elements – the drug product itself; the primary pack, such as a PFS with a staked needle or a 3 mL cartridge; and a delivery system such as an autoinjector. The shift in collaboration is driven by the complex interdependencies between the drug, its primary packaging (such as a PFS or cartridge) and the delivery device. It demands a nuanced approach.
The form and function of the delivery device can affect the way that the drug itself is formulated and vice versa. Consideration should be given to how the drug will interact with the device to ensure consistent injections. Each device component must be examined and tested for efficacy in combination with other parts. Chemical and physical stability, device compatibility and patient usability must be balanced. PFSs, for example, expose drug products to prolonged contact with elastomeric components and lubricants, complicating the delivery of uncompromised quality in the drug formulation.
In the past, this interaction between the drug and the delivery system received insufficient early attention. As shown in Figure 1, working in silos only to bring the device and drug together at a later stage can result in unexpected incompatibilities that send a team back to the drawing board with significant delays, added costs and rework.
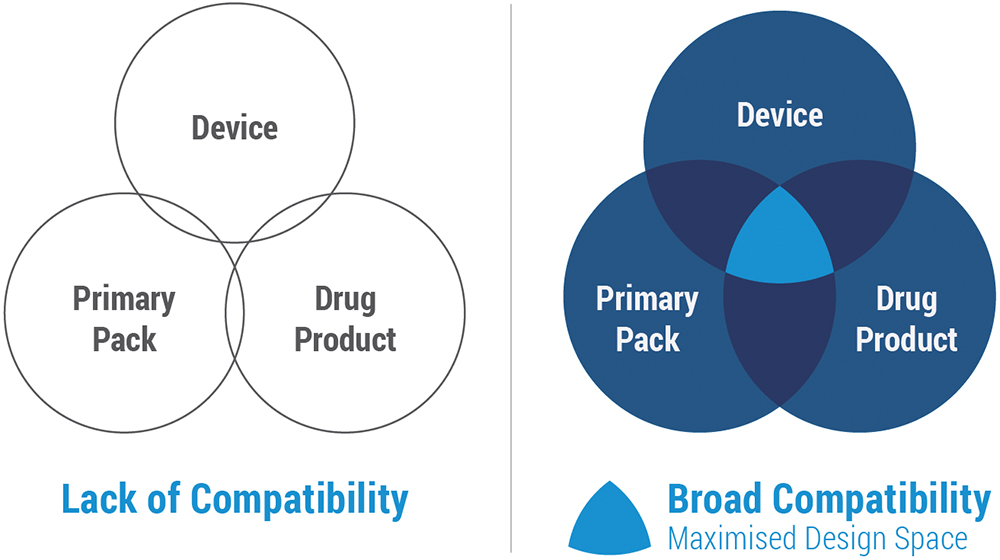
Figure 1: Compatibility among components.
Formulation science is broad, and there is not necessarily one formulation that will work for a drug. However, once clinical trials have begun and formulation efficacy data are gathered, it becomes increasingly difficult for the drug maker to change it without added time, costs and complications.
Considerations for drug-device compatibility include the following:
- Lubricants used in primary packs (such as PFSs) can impact drug stability
- Excipients may cause an unexpected reaction
- Stabilisers, such as sucrose and trehalose, can increase viscosity and impact injection force
- Buffers must be compatible with syringe materials to minimise risks such as aggregation
- Surfactants prevent surface adsorption but may degrade into particulates, potentially clogging needles and disrupting reliable dose delivery.
Device engineers must accommodate formulation challenges to account for the delivery system. Table 1 shows the sensitive interdependencies that exist between each of the components. In almost all cases, the critical quality attributes rely on two or more elements working together synergistically. Dose delivery and injection time rely on all elements – formulation, primary pack and injector – working together.
| Critical Quality Attributes | Formulation | Prefilled Syringe | Autoinjector | Manufacturing |
| Identity | High | None | None | None |
| Assay/Impurity | High | Weak | None | Weak |
| Sterility | Weak | Weak | Weak | |
| Particulate Matter | High | High | None | |
| pH/Osmolality | High | Weak | None | Weak |
| Viscosity | High | High | High | High |
| Dose Delivery Accuracy | High | High | High | High |
| Injection Time | High | High | High | High |
| Activation Force | None | None | High | Weak |
Table 1: Interdependence of key components.
There has been a significant shift in how drug and device development intersect over the past few years. It is notable that in recent years, clients with drugs in preclinical stages, not yet undergoing IND-enabling studies, are enquiring about autoinjectors types, options available and multidose flexible patterns.
When drug and device developers assess interactions and key factors together, they will have more flexibility to make modifications and solve adverse interactions. Executing this during early phases before generating clinical data can prevent design rework on the back end.
HUMAN FACTORS AND USABILITY
The patient is at the heart of modern injectable drug development. Usability testing, human factors studies and real-world data collection are now integral to the process. Regulatory agencies expect manufacturers to demonstrate ease of use, accessibility and minimal burden on the patient – in addition to clinical efficiency – during submissions.
Formative human factors studies are valuable early on and throughout the development programme. This includes both the drug delivery system and the proposed instructions for use. Comprehensive training programmes, such as those implemented for prefilled pen devices in clinical trials, help to ensure that patients use correct injection techniques, reducing variability in trial data and accelerating time-to-market.
Fewer than 14% of drugs progress from Phase I trials to the commercial market.1 Recent FDA transparency initiatives show that usability gaps, such as missing human factors validation, are increasingly cited in Complete Response Letters (CRLs) for combination products. These deficiencies often trigger requests for additional studies, delaying approval.2 CRL issuance and resubmission cycles can add median delays of ~435 days or a range from 47 up to 2,374 days before eventual approval.3
It is important that, if a particular route of administration with a drug is not performing as expected, it is identified as early as possible to minimise wasted investment. It is equally important that drug assets with demonstrated value are made available to patients as early as possible.
CLINICAL TRIAL DESIGN
Designing clinical trials is crucial for the commercial success of a new drug. Integrating formulation properties with delivery device design early in the drug development process provides significant long-term advantages. Matching device capability with formulation properties is key to developing robust, differentiated combination drug products.
As pharma companies consider the interplay between the regulators, contract research organisations, study sponsors and themselves, it is important that each new drug has the best opportunity to succeed through collaborative processes. This seems to be industry’s direction of travel, with smart technologies and artificial intelligence increasingly being introduced into clinical trials to collect data and achieve results as quickly as possible.
Connected devices can enhance clinical trial speed and assessments of drug performance by automatically monitoring and reporting data on self-administration practices, compliance and device performance. Connected autoinjectors, if designed well, remove reliance on patient self-reporting and provide ongoing feedback that informs future device improvements. However, human factors must also be tested with any added technology. Imposing added technological burdens – such as requiring less digitally savvy populations to input data into a smartphone app – may not be the best route when more passive connectivity methods are possible.
Passive data transmission, such as autoinjectors that use a charging station for data capture and automatically send usage data via a cellular network without any patient action, supports decentralised trials in more remote, less populated regions.
REGULATORY STRATEGY: DATA IS KEY
Navigating the regulatory landscape for combination products is complex. Agencies expect manufacturers to demonstrate not only clinical efficacy but also usability, safety and accessibility. While the FDA is centralised and the EMA is decentralised with additional steps for device conformity, they both emphasise usability, risk management and integrated lifecycle controls. Early engagement with regulatory bodies and comprehensive data generation on device compatibility, stability and human factors are essential.
Processing data becomes much easier when healthcare providers can see the frequency of dosing and how patients are navigating their treatment protocols. Clinical endpoints can be affected by patient deviation from protocol-defined adherence, compliance and persistence targets. Connected devices that automatically upload data in real time provide faster and more reliable data, which can help to address issues with a drug product that may have suffered a setback or even a failure because of patient adherence problems. Earlier and more frequent data collection enables issues to be addressed sooner, leaving teams better equipped to resolve problems without having to repeat studies or extend trials because the data are late or do not make sense.
Addressing the success factors listed in Figure 2 requires co-operation between formulation scientists and device engineers. Delivering high-viscosity formulations demands precise co-ordination of needle design, drive-force mechanics and fluid resistance dynamics to balance injection time and patient comfort. Usability studies and patient feedback then help to refine these systems to improve ease of use and overall device performance.

Figure 2: Success depends on viscosity, dose accuracy and injection time.
CONCLUSION
The shift towards patient self- administration of injectable drugs is reshaping the pharmaceutical industry. Collaboration between drug and device developers has become a strategic advantage, allowing the device to be customised for the specific requirements of the drug product while ensuring desired performance and reliability. The benefits include minimising design cycles, shortening development timelines and reducing associated costs.
By embracing cross-disciplinary co-operation, integrating human factors and usability studies, and engaging regulatory bodies early, pharma companies are better able to deliver therapies that are safe, effective and accessible. This integrated approach offers a smarter, faster and more patient-aligned path to both clinical and commercial development. In an increasingly competitive landscape, it is the key to improving patient outcomes and achieving market success.
REFERENCES
- Cross R, “Drug development success rates are higher than previously reported”. Chemical & Engineering News, Feb 2018.
- Waldron J, Kansteiner F, “‘Weak’ evidence and an ‘unpleasant’ odor: FDA sheds light on drug refusal process”. Fierce Biotech, Jul 2025.
- “Top Reasons FDA Rejects Drug Applications: Lessons from Safety and Adverse Event Data”. Velsafe, Aug 2025.