

To Issue 156
Citation: Scatena A, Toth S, Chenevert K, “Exploring Extractable and Leachable Testing Strategies for Parenterals”. ONdrugDelivery, Issue 156 (Jan 2024), pp 102–106.
Antonio Scatena, Scott Toth and Kyle Chenevert discuss the regulatory landscape regarding extractables and leachables for parenteral drugs and provide guidance on implementing a successful testing approach. Techniques and results interpretation are discussed, emphasising practical advice for developers working towards regulatory submission.
Contamination of a drug by extractables and leachables (E&Ls) can lead to drug recalls and significant commercial losses, as evidenced by a major recall of children’s medicine in 2010 (McNeil Consumer Healthcare, PA, US) linked to the migration of 2,4,6 -tribromoanisole from shipping pallets,1 and a recall of felodipine (Mutual Pharmaceutical, PA, US) due to benzophenone leaching from varnish on the packaging.2
In some cases, E&Ls can also result in adverse effects, as was observed in the early 2000s with the spike of pure red blood cell aplasia (PRCA) cases that were linked to the use of injected erythropoietin (EPO). In this case, rubber extractables were identified as the leading cause of EPO aggregate formation,3 which may have elicited auto-immune responses in patients and resulted in PRCA.4
Examples like these highlight the critical role of E&L testing in ensuring drug safety, particularly for dosage forms such as injectables. These drugs fall into the US FDA’s highest risk categories, as they are directly injected into the parenteral space. Furthermore, the likelihood of interaction between dose and packaging is high for liquid-state drugs.5
Extractable testing reveals compounds released by a given material under relatively aggressive conditions, while leachable testing seeks to detect and quantify the migration of compounds into a specific drug product formulation under representative storage conditions (Figure 1). Both are subject to extensive regulatory guidance and evolving industry practice, none of which is truly prescriptive. Instead, drug developers must demonstrate a rigorous and systematic approach that identifies problematic compounds for a specific drug product and minimises risk by ensuring their effective control.

Figure 1: Extractables are compounds that could migrate into the product; leachables are compounds that migrate into the product under representative conditions.
“Drug developers must demonstrate a rigorous and systematic approach that identifies problematic compounds for a specific drug product and minimises risk by ensuring their effective control.”
EXTRACTABLES & LEACHABLES: SOURCES AND RISK
Primary packaging is a defining focus when it comes to E&L testing for parenterals, and rightly so, since it is not unusual for a ready-to-use drug product to be stored for up to 24 months. Solutions and suspensions for parenteral delivery are typically filled into vials, bottles and syringes made from glass or cyclo-olefin polymer or co-polymer (COP/ COC) and closed with rubber components. Rubber manufacturers have developed various strategies for rubber lamination (such as ethylene tetrafluoroethylene film coating), which aims to limit the risk of compound migration into the drug, further emphasising the focus on E&L risk mitigation for primary packaging.
For some applications, solutions can be packaged in flexible containers made from polymers, such as polyethylene or polyvinylchloride (PVC). The associated container closure systems (CCSs) are particularly important with respect to leachables, not least because of their steadily evolving complexity. The growing use of CCSs with a multi-layered construction, incorporating plastics, adhesives and foils, has contributed to increased scrutiny of these components regarding the potential for releasing E&Ls ranging from pigments to plasticisers.
Primary packaging is the main source of E&Ls for parenteral drugs, but other sources, such as manufacturing componentry and secondary packaging, may also need to be considered (Figure 2). As a drug product advances through the manufacturing chain, it contacts various surfaces and materials, from vulcanised rubber tubing to lubricants and metal alloys. Typically, contact times are short, but not always, with processes such as filtration providing opportunities for extended interaction. While secondary packaging may be further down the list of likely causes of contamination, it can be a fruitful avenue of investigation when alternative sources have been eliminated.
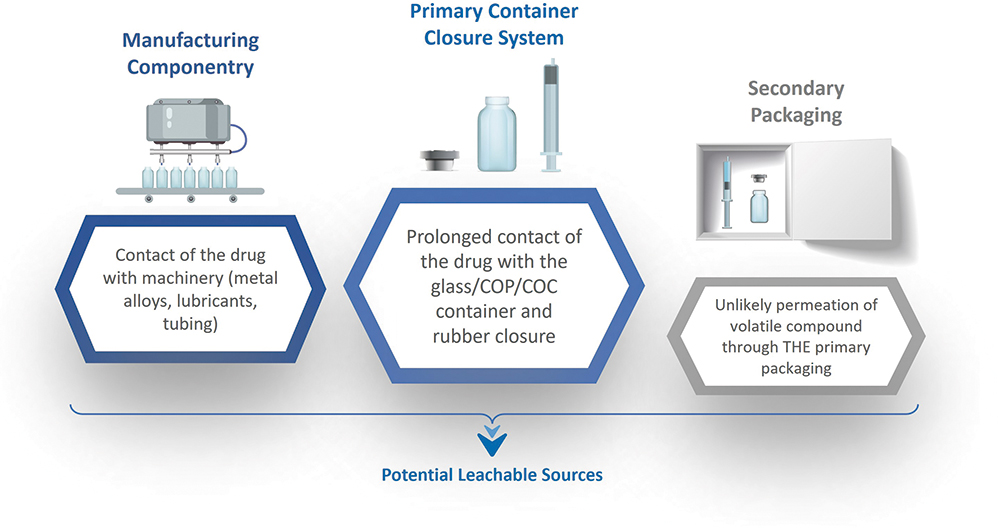
Figure 2: Sources of potential leachables include manufacturing componentry in contact during production, primary packaging, the container closure systems and secondary packaging.
Patient safety is the primary concern regarding E&Ls because migration into the drug product can result in the delivery of toxic impurities along with the API or adversely affect its stability and potency.3
However, results from extractables or simulation studies are also helpful to drug developers when selecting packaging materials to preserve the long-term quality of the drug and maximise drug stability. For all parenteral drugs, but particularly for sensitive biologics such as monoclonal antibodies, recombinant peptides or mRNA vaccines, E&Ls may trigger aggregation, unfolding and/or oxidation, thereby limiting shelf life, compromising therapeutic efficacy and, in the worst case, affecting patient safety.3-4
“When designing a study, the first question is whether the scope includes both E&Ls or whether the focus is one or the other.”
THE REGULATORY LANDSCAPE AND CURRENT BEST PRACTICE
The FDA advocates a risk-based approach to E&Ls for parenterals, emphasising the need for developers to minimise adverse patient impacts through systematic study and assessment.5 The absence of prescriptive protocols highlights the benefits of working with experts in the field, although there is published guidance to reference, notably from the Product Quality Research Institute(PQRI).6 Released in 2021, the latest PQRI guidance is especially helpful for clarity around the safety concern threshold (SCT) concept. Defining the SCT “as the threshold below which a leachable would have a dose so low as to present negligible safety concerns from carcinogenic and noncarcinogenic toxic effects” suggests an SCT of 1.5 μg/day as suitable for the majority of organic leachables in parenterals. This figure helps to define the sensitivity with which to look for migrating compounds. Insights from the FDA are also helpful in this regard, indicating that, for parenterals, the common practice is to report leachables at > 1 ppm, identify them at 10 ppm and qualify them at 20 ppm.5
US Pharmacopoeia (USP) <659> covers packaging and storage requirements with the following opening statement “Packaging materials must not interact physically or chemically with a packaged article in a manner that causes its safety, identity, strength, quality or purity to fail to conform to established requirements.” Individual chapters specifically refer to commonly used packaging for pharma products – USP <381> for rubber closure, USP<660> for glass and USP <661> for plastic packaging. For rubber closure and plastic containers, extractables are specifically addressed in USP <1663> and leachables in USP <1664>, which both specify the test methods that need to be implemented for adequate evaluation.
Change is also underway regarding International Council of Harmonisation (ICH) guidance, with the much-anticipated ICH Q3E Guideline for Extractables and Leachables currently being finalised and expected to be adopted by 2026. The ICH M7 guideline, which relates specifically to mutagenic compounds, remains in place and is helpful for reference but, in practical terms, has been largely superseded by the PQRI document.
DESIGNING A STUDY
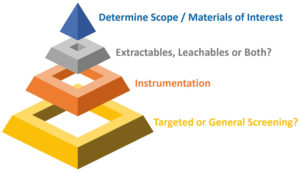
Figure 3: An E&L study requires careful consideration of several factors.
For effective study design, it is necessary to clearly differentiate E&L testing and understand how the two relate to one another. Extractable testing aims to identify a profile of compounds that could be released from a material used for packaging or manufacturing componentry by applying relatively harsh – though somewhat representative – conditions. By “pushing” candidate materials, extractable testing effectively determines a worst-case leachable profile. The results help to qualify alternative options for CCSs, establish quality control protocols for material acceptance/use and screen for the release of toxic materials. Extractable testing is often a proving ground for analytical techniques destined for application in leachable testing. In contrast, leachable testing determines which compounds migrate into a specific drug formulation under closely representative conditions and, as a result, is much more akin to stability testing.
When designing a study, the first question is whether the scope includes both E&Ls or whether the focus is one or the other. Additional questions that are usefully answered at the outset include the following (Figure 3):
- What materials are of interest from the perspective of primary packaging, notably the CCS and manufacturing componentry?
- Are there specific compounds of concern, or is the intention to perform a broad screen? A broad screen will deliver an unbiased assessment of any compounds present, but sensitivity can be as much as five times greater for a targeted study.
- What analytical techniques and instrumentation are going to be relevant?
- What conditions are going to be appropriate for testing?
Consider this last point in greater detail by examining a general workflow for extractables testing (Figure 4).
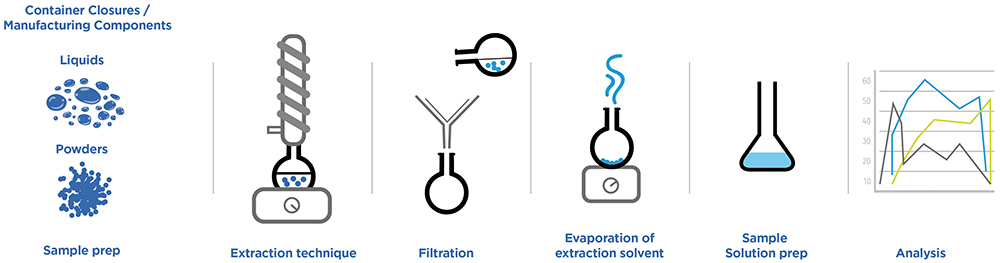
Figure 4: An execution flow for extractable testing highlights the need to consider various factors when developing an appropriate test strategy.
“The first step in processing and interpreting E&L test data is to identify compounds of interest .”
Assessing each of these steps highlights the wide range of factors to consider when developing an extractable test protocol to apply sufficiently harsh conditions while remaining within the boundaries of remaining realistic. Some critical questions to address are:
- What form should the test sample take? Would there usually be any form of pre-treatment, such as washing or irradiation, that should be reflected in the test protocols?
- Which extraction technique(s) is most appropriate – microwave, reflux, Soxhlet, sonication or accelerated solvent extraction?
- Which solvents should be used – polar (e.g. water, isopropanol) or non-polar (e.g. dichloromethane, hexane), a range or a combination? Are additives needed to, for example, assess the impact of pH and/or surfactants?
- What are the appropriate contact time and temperature ranges? Temperature tends to be in the range of 25°C–40°C, with test times extending from a few hours up to 70 days.
- Is the accessible contact surface area suitable? A surface area-to-volume ratio of 6:1 cm2/mL is routinely adopted.
- What will be the negative control? In other words, which highly inert materials – high-grade borosilicate glass and polytetrafluoroethylene are common candidates – will be used to establish a baseline for comparative purposes?
- Is there an appropriate internal standard to track the efficiency of the sample work-up process – usually a molecule of comparable class/structure to those of interest?
- Does the sample require cleaning up to improve the quality of analytical data via, for example, filtration or concentration?
- Is further concentration required to reach the compound’s analytical evaluation threshold (AET)? Possible concentration techniques include evaporation, liquid–liquid extraction, dispersive liquid–liquid microextraction, hanging droplet microextraction and solid-phase microextraction.
- Which analytical technique is most appropriate? Liquid chromatography ultraviolet mass spectrometry (LC-UV-MS) is valuable for non-volatile compounds, including oligomers and larger antioxidants; gas chromatography – MS (GC-MS) with liquid injection is useful for semi-volatile compounds, such as residual monomers, preservatives and plasticisers, with GC-MS for volatile compounds via headspace analysis better suited to inks, adhesives and process solvents; and inductively coupled plasma MS (ICPMS) useful for elemental analysis, notably metals.
This is a lengthy list of considerations, and many are equally applicable to leachable testing, highlighting the considerable effort involved in implementing an effective study. It is also important to recognise the value of standardisation in extractable testing, as it supports the effective comparison of alternative materials and broader use/sharing of extractable testing data, thereby reducing the associated workload. Information released by the BioPhorum Operations Group7 is helpful in this regard and usefully referenced. For leachables, on the other hand, every drug product is unique, and individually tailored studies are therefore required.
PROGRESSING TO ACTIONABLE INSIGHT
The first step in processing and interpreting E&L test data is to identify compounds of interest. These may be targeted molecules associated with using certain materials or unknowns absent from the original drug formulation and negative control. For the latter, routes to identification include commercial MS libraries, molecular formula generation from empirical mass/isotope spacing on a high-resolution instrument and/or MS/MS fragmentation in combination with resident time matching with an authentic standard.
Having identified compounds of interest, the next step is determining the AET – “the threshold at or above which a chemist should begin to identify a particular leachable and/or extractable and report it for potential toxicological assessment”.6 The AET brings specificity to the SCT, essentially converting it into a metric that takes account of how the drug will be used, since this, in turn, determines the extent of patient exposure (Figure 5). The administration method, the concentration of drug product, the amount per dose, dosage frequency and dosage duration all impact the AET, which is set to ensure adequate patient protection across intended use scenarios. For example, the AET for a vaccine delivered two to three times across a patient’s lifetime will be significantly higher than that for an identical leachable detected in a parenteral for daily dosing to treat chronic disease. ICH M7 helps determine AETs, specifically for mutagens, and values the testing strategy in terms of stringency and precision. The lower AETs associated with a routinely used drug may necessitate the further refinement of test strategies to enhance sensitivity to the required level.
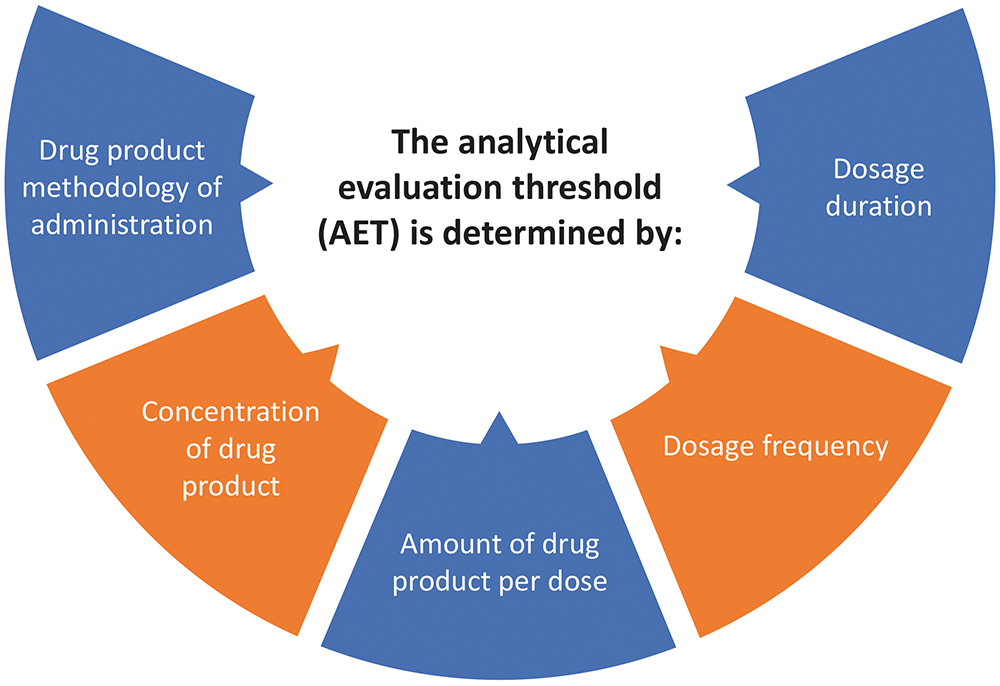
Figure 5: The AET is a critical concept in E&L testing, determined from consideration of the way in which the drug will be used.
Ultimately, the outcome of a successful E&L study is a profile of the identity and concentration of all compounds of concern, their source (if known) and any additional data associated with potential risk to the patient. This can be used to make a toxicological assessment for the product, greenlighting further development or prompting modification. In this way, E&L studies form an integral and critical element of parenteral development and the associated regulatory submission. Ensuring that all materials that the drug product contacts throughout its lifetime are appropriate for use is a cornerstone of drug safety and an area of growing scrutiny, particularly for exacting, higher-risk products such as parenterals.
E&L studies are unique for each new drug and represent a significant forensic and analytical chemistry exercise. Working with experts can be the way forward for those new to the area or daunted by the challenge. Developing in-house abilities to successfully design and execute these vital studies requires significant investment and resources. Getting help can be highly productive in getting safer parenterals to market faster.
REFERENCES
- Lynch M P, “Extractables and Leachables Quality Concerns and Considerations for Ophthalmic and Injectable Products.” Am Pharm Rev, May 2011.
- Palmer E, “Sun Pharma recalls more than 216,000 bottles after label leaches chemical”. Fierce Pharma, Sep 2015.
- Richter C et al,”Impact of extractables from rubber closures on protein stability under heat stress”. Eur J Pharm Biopharm, 2018, Vol 130, pp 22–29.
- Sharma B, “Immunogenicity of therapeutic proteins. Part 2: impact of container closures”. Biotechnol Adv, 2007, Vol 25(3), pp 318–324.
- Lewis DB, “Current FDA Perspective on Leachable Impurities in Parenteral and Ophthalmic Drug Products”. AAPS Workshop on Pharmaceutical Stability – Scientific and Regulatory Considerations for Global Drug Development and Commercialization, Oct 2011, Washington, DC.
- “Safety Thresholds and Best Demonstrated Practices for Extractables and Leachables in Parenteral Drug Products (Intravenous, Subcutaneous, and Intramuscular)”. Product Quality Research Institute, Oct 2021.
- “Disposables: Extractables testing of polymeric single-use components used in biopharmaceutical manufacturing”. BioPhorum, accessed Dec 2023.
Previous article
A NOVEL INSTRUMENT TO STREAMLINE COMBINATION PRODUCT DEVELOPMENTNext article
Interview with Lilli Zakarija, EdgeOne Medical
