Citation: Patel A, “The Path to Commercialisation for Wearable Drug Delivery Devices”. ONdrugDelivery, Issue 124 (Sep 2021), pp 58–62.
Atul Patel looks at the evolution of the wearable drug delivery market, including the route to commercialisation and the challenges faced.
“For patients, wearables offer the convenience of taking medication at home, avoiding the time and effort of visiting a clinic, thereby improving quality of life.”
Wearable medical devices have transformed healthcare. Ever since the US FDA granted Cygnus (ID, US) approval to market for its GlucoWatch Biographer as a prescription device for adults with diabetes in 2001,1 the wearables market has continued to evolve and now includes a variety of wearable injectors – devices designed to deliver drugs in large volumes subcutaneously.
Some analysts predict that the global wearable injectors market will reach US$18.3 billion (£13.2 billion) by 2028.2 This growth is being driven by the benefits these devices deliver to various stakeholders. For pharmaceutical and biopharmaceutical companies, wearables offer the opportunity to grow market share and reduce competition. For payers and providers, wearables can reduce costs associated with in-clinic treatment and, potentially, improve outcomes due to better medication adherence. And for patients, wearables offer the convenience of taking medication at home, avoiding the time and effort of visiting a clinic, thereby improving quality of life.
The home care end-use segment of the wearables market grew significantly in 2020, reflecting a preference for self-administration that was growing even before the covid-19 pandemic. In a 2018 study conducted in Belgian hospitals, for instance, the majority of patients surveyed said they would prefer to self-administer medication, and they had a positive attitude towards self-administration of medication.3
To capitalise on this trend, the pharmaceutical and biopharmaceutical industries have responded by introducing next-generation therapies that combine a medicine with a delivery system conducive to self-administration. These combination products, as designated by the FDA, can take several different configurations: a drug and a device; a biologic and a device; a drug and a biologic; or a drug, a biologic and a device. These products also include combinations that are physically, chemically or otherwise mixed and produced as a single product; two or more products contained in a single package; or separate products that are cross-labelled to be used together.4 For most wearable injectors, their designation as a combination product seems clear, but even a prefilled syringe – a relatively simple drug-device combination – is viewed by the FDA as a combination product. It should be no surprise, then, that combination products account for approximately one-third of all medical products under development today.
What is not apparent in this statistic are the enormous challenges that must be overcome when commercialising any combination product. A more typical approach – one in which early development focuses on the drug, leaving product and device considerations to later phases – introduces significant uncertainty and risk. For combination products, such as a wearable injector, the drug development process and the device development process must come together at the earliest stages to help guide the overall product strategy and to meet FDA expectations from a compliance standpoint. Considering the drug and device separately can introduce significant risk, even at the earliest stages of a project. Figure 1 shows what a more integrated approach might look like.
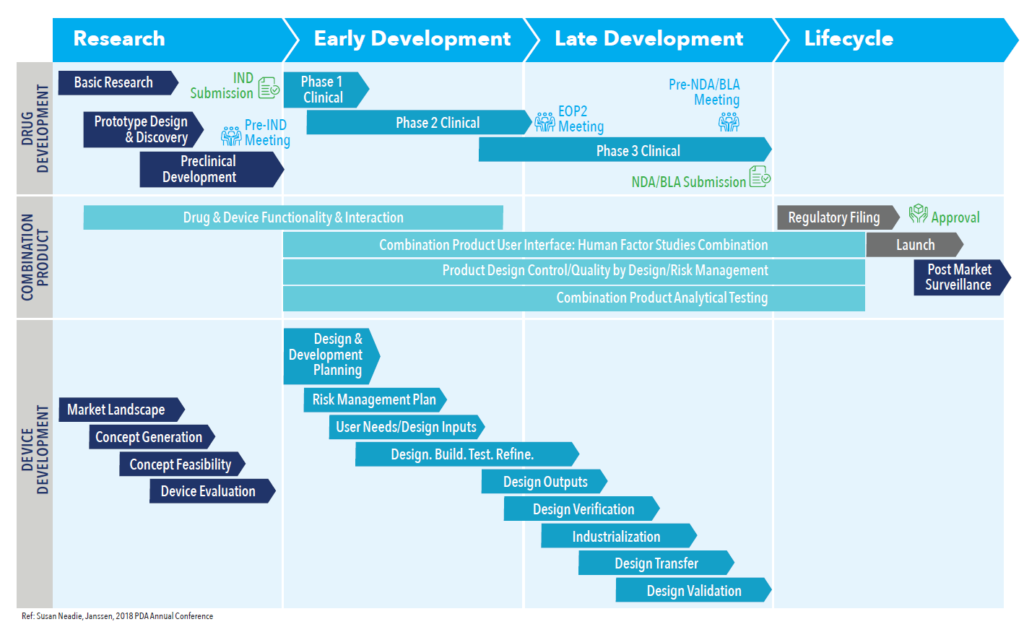
Figure 1: The drug development process and device development process come together as early as possible.
“For combination products, such as a wearable injector, the drug development process and the device development process must come together at the earliest stages to help guide the overall product strategy and to meet FDA expectations from a compliance standpoint.”
A two-pronged development process facilitates planning throughout the product life cycle and enables the sponsor team to address the following key challenges and considerations that can stand in the way of commercial success:
- Navigating the complex regulatory landscape for combination products
- Choosing the right presentation for the drug at the appropriate time
- Managing supply chain complexities.
REGULATORY CHALLENGES
Developing a device and drug in parallel requires synergies and collaboration among a number of concurrent workstreams, and they must answer a series of interconnected questions:
- What is the product’s primary mode of action (PMOA)?
- Is the drug compatible with the containment system?
- Is the containment system compatible with the fill-finish process?
- Is the containment system compatible with the delivery device?
- Has the device been designed with both manufacturability and end-user needs in mind?
Making the wrong choice at any critical juncture or with any critical component can result in delays and costly reworks, but answering these questions with confidence is daunting because of the mercurial nature of the regulatory landscape. Since the final rule on current good manufacturing practice (cGMP) requirements for combination products (21 CFR Part 4) was issued in January 2013, there have been many draft guidances and other regulations introduced. It is not always easy to understand the impact of new regulations – especially when moving into different geographic regions – but failing to provide the necessary proof and documentation can result in a rejected filing, delaying the time to market.
To ensure compliance with regulatory requirements, human factors and analytical testing must be fully integrated into the development process. Human factors testing explores how end users interact with a device, and the FDA guidance focuses on three aspects: the intended users, the use environments and the user interface.5 Regulators look for evidence of safe and effective use of a product from these three perspectives.
Analytical testing is particularly complex for combination products. Development teams must produce data to demonstrate compatibility, functionality and performance to support regulatory requirements. As ISO guidelines supporting the drug-device combination continue to evolve, however, this can be difficult. For example, the ISO 11.040 standards govern medical equipment as a category – standard 11.040.25, which provides guidance for syringes, needles and catheters, includes 73 standards alone, all or some of which may apply to a combination device. Additional testing requirements could also include bridging studies. These tests may include the bridging of data from combination products that employ different device components for the same drug or biologic, as well as the same device component across different drugs and biologics. Bridging study data are typically provided in addition to the foundational information included in an IND, BLA or NDA.
“Choosing the right way to present a combination product and collecting the right data is complex. Having a development roadmap from the outset of a project is critical to build a clear picture of the requirements needed throughout all phases of development.”
PATHWAYS AND PRESENTATION
Interacting with regulatory bodies presents its own unique challenges. Central to this is determining which type of premarket submission is appropriate. With the FDA, the agency taking the lead to evaluate a combination product is determined based on which constituent part of the product provides the PMOA. There are three primary regulatory pathways a product can follow: a device-led combination product, a drug-led combination product or a biologic-led combination product.
The FDA typically only requires a single application, regardless of pathway, which eliminates unnecessary duplication that may occur with multiple applications. However, there may be times when a developer decides to submit multiple applications (e.g. new drug product exclusivity, orphan status or proprietary data protection when two firms are involved). In these scenarios, the individual constituent parts of the combination product will be reviewed by applicable centres.
To determine the safety and efficacy of the whole combination product, the FDA will consider regulations for each constituent part. For a device-led product, that includes a drug constituent – the FDA will need to see data on the drug, including nonclinical and clinical pharmacology information and chemistry. This data for the non-constituent part of a combination product are not necessarily the same safety and effectiveness data required for the same part being submitted as a stand-alone drug.
Clearly, choosing the right way to present a combination product and collecting the right data is complex. Having a development roadmap from the outset of a project is critical to build a clear picture of the requirements needed throughout all phases of development. It is also important to look for partners who can supply components with technical data, which makes it far easier to provide reliable data in support of development decision-making.
SUPPLY CHAIN COMPLEXITY
The ever-changing regulatory, analytical and manufacturing needs of combination products leads to another significant challenge – supply chain complexity. Consider the scenario in which a pharmaceutical company, an expert at traditional drug development, must now think about the design of a delivery device, yet lacks in-house design expertise. The design of a device must be challenged to ensure that it does not interact with the drug. At the same time, a development team must also consider the integrity of the container closure, as well as the stability and extractables/leachable profiles.
To accommodate these design needs, a pharmaceutical company will typically outsource the work to a contractor, usually just one of many. In fact, many companies stitch together an ecosystem of multiple vendors, typically with each vendor operating independently. This can result in material shortages, inaccurate lead-times, distribution bottlenecks, quality control issues, failed inspections and lack of documentation.
Instead of this, it is preferable to work with a single partner that brings together both extensive knowledge of product design and development and the support services necessary to mitigate risk, reduce regulatory complexity and simplify the supply chain. Figure 2 shows all of the integrated solutions required to bring a combination product to the market. Clearly, having a development partner that offers some or all of these services under one roof – especially a partner with extensive combination product experience – can help to mitigate obstacles, avoid pitfalls and accelerate time to market.
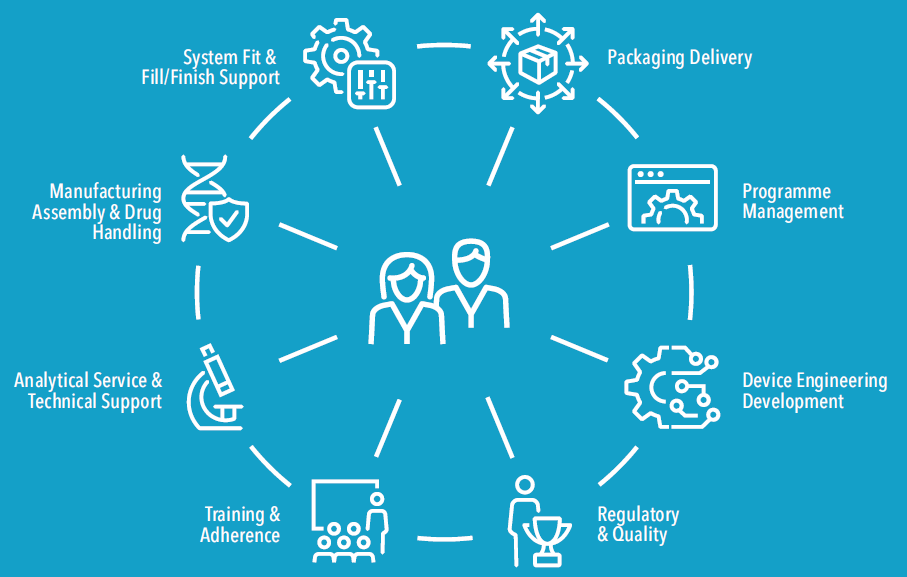
Figure 2: Product development teams often rely on a complex network of vendors to supplement their in-house capabilities, but a better solution is to find a single partner with extensive combination product experience.
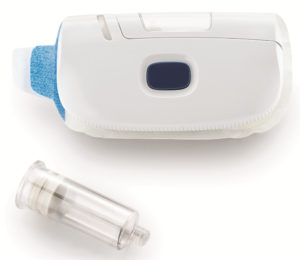
Figure 3: West’s SmartDose® 10 Injector delivers a dose volume of up to 10 mL, which makes it suitable for next-generation, high-viscosity drug products.
CASE STUDY: WEST’S SMARTDOSE® 10 INJECTOR
West Pharmaceutical Services, a leader in the design and production of integrated containment and delivery systems for injectable medicines, has extensive real-world experience bringing combination products to the market. The company partners with the world’s top pharmaceutical and biotechnology companies to bring technologically advanced, patient-centric, high-quality products to the market.
West’s SmartDose® 10 Injector (Figure 3) stands as an ideal example of what these partnerships can produce. The next evolution of the SmartDose® 3.5 Injector, which was approved by the FDA in 2016 for delivery of Amgen’s (CA, US) Repatha® (evolocumab) drug treatment, the SmartDose® 10 Injector delivers a dose volume of up to 10 mL, which makes it suitable for next-generation, high-viscosity drug products. Typically, such drugs would be administered in clinical settings, creating additional burden for the patient. The SmartDose® 10 Injector eliminates these issues by enabling self-administration of oncology and neuroscience therapies at the patient’s home. The device is accompanied by detailed onboarding and training, and offers visual, tactile and audible feedback to boost user confidence. The result is an easy-to-use, patient-centric solution that eases patient anxiety and increases adherence.
Because of both these benefits and West’s experience with commercialising wearable injectors, the SmartDose® 10 Injector has seen increasing market acceptance. For example, in 2019, scPharmaceuticals (MA, US) announced its intent to go to market with West’s SmartDose® 10 injector for FUROSCIX®, a proprietary, subcutaneously delivered furosemide solution for the treatment of worsening heart failure due to congestion. In July 2021, scPharmaceuticals announced that it is moving forwards with required bench testing and, subject to the completion of the Drug Master File by West, it is targeting the resubmission of its NDA in the fourth quarter of this year with an anticipated six-month review by the FDA.6
“As therapies become more complex and shift towards self- administration, pharmaceutical and biopharmaceutical manufacturers are under pressure to bring more intricate devices to the market that are also easy to use.”
SIMPLIFY THE JOURNEY™ WITH WEST
As therapies become more complex and shift towards self-administration, pharmaceutical and biopharmaceutical manufacturers are under pressure to bring devices to market that are both more intricate and easy to use. This is particularly challenging when constantly changing regulations place more responsibility on sponsors to prove not only that their drug is reliable and safe but also that the device is fully integrated and satisfies usability requirements. Relying on a provider with both combination product and services expertise allows manufacturers to simplify their supply chain and gain compelling benefits, such as fewer partners, improved knowledge transfer and scalability across a product’s life cycle, and faster root cause analysis and resolution if problems arise.
West works as a true partner with its pharma and biopharma customers, and has supported multiple development programmes through its trademarked “Simplify the Journey” process. All of this experience adds up to an impressive résumé defined by a range of capabilities.
West offers:
- Leadership in testing and verification of complex injectable drug containment, delivery systems and combination products, with the ability to support design validation testing.
- An extensive resource knowledge centre built on over 90 years of experience in developing, testing, manufacturing and commercialising containment systems and devices.
- Proven containment and device design and development history, including experience taking products from concept to commercialisation.
- A seasoned team, including human factors, regulatory and device development experts.
- A breadth of capabilities and deep expertise to support combination products from preclinical to post-market launch.
- Integrated fill-finish services ranging from small-scale fills suitable for the clinic to commercial-scale production volumes.
- Over 50 years of contract manufacturing experience, including drug delivery devices, diagnostics and medical devices.
- A platform approach to shorten time to market.
West leverages these capabilities every day on behalf of the world’s top pharmaceutical and biotechnology companies, and can help development teams navigate the challenges and mitigate the risks encountered on the path to commercialisation. West offers a full portfolio of vial container closure system solutions, as well as self-injection platforms, such as the SmartDose® drug delivery system, that can help accelerate product development. West supplements this offering with a suite of analytical services, contract manufacturing capabilities, regulatory guidance and a cadre of professionals dedicated to de-risking the transition to combination product – and developing patient-centric solutions that improve the health and quality of life of patients.
SmartDoseV is a registered trademark of West Pharma. Services IL, Ltd., a subsidiary of West Pharmaceutical Services, Inc. Simplify the Journey™ is a trademark of West Pharmaceutical Services, Inc. RepathaV is a registered trademarks of Amgen Inc. FUROSCIXV is a registered trademark of scPharmaceuticals Inc.
REFERENCES
- “FDA approves first non-invasive, automatic glucose monitoring system for people with diabetes”. Press Release, Med Device Online, Mar 2001.
- “Wearable Injectors Market Size Worth $18.3 Billion By 2028 | CAGR: 17.2%”. Press Release, PR Newswire, Mar 2021.
- Vanwesemael T et al, “The willingness and attitude of patients towards self-administration of medication in hospital”. Ther Adv Drug Saf, 2018, Vol 9(6), pp 309–321.
- “Combination Product Definition Combination Product Types”. FDA, Feb 2018.
- “Draft Guidance for Industry and FDA Staff: Human Factors Studies and Related Clinical Study Considerations in Combination Product Design and Development.” US FDA, Feb 2016.
- “scPharmaceuticals Inc. Announces Receipt of Written Minutes from Type C Meeting with the FDA Regarding Development of FUROSCIX®”. Press Release, scPharmaceuticals, Jul 2021.