Citation: Gray R, Rashba-Step J, “Automated Lipid Nanoparticle Production: from Protocol Development to GMP Manufacture”. ONdrugDelivery, Issue 123 (Aug 2021), pp 48–52.
Richard Gray and Julia Rashba-Step discuss the benefits of automated lipid nanoparticle production.
“Current nanoparticle production methods tend to either be batch based or use relatively large-scale, continuous flow equipment, such as membranes. Both methods have significant drawbacks.”
Interest in nanotechnology, including both polymeric and lipidic nanoparticles, has grown significantly in the last decade. The tremendous success of mRNA-lipid nanoparticle (LNP)-based vaccines during the covid-19 pandemic has led to further explosive growth in development and created new opportunities in this area. The unprecedented speed of development of the covid-19 LNP-based vaccines was, to a large extent, built on groundwork performed over the last 10–15 years in LNP platform development.
Nanoparticles are excellent delivery vehicles for various modalities, including nucleic acids such as mRNA, saRNA and siRNA, proteins and small molecules, as well as non-pharmaceutical products in the food, cosmetics and other industries. The function of the nanoparticle is to deliver the cargo to the action site and to protect the cargo – often referred to as an API – from degradation within the body and during storage.
Lipidic particles, including liposome and lipid nanoparticles, are spherical vesicles of various sizes (typically ranging from 30 to several hundred nanometres) and architecture, formed via self-assembly. These lipidic structures have the capacity to encapsulate both hydrophobic and hydrophilic compounds, and offer increased bioavailability and targeted delivery. To enhance the desired properties, it is feasible to functionalise the particle surface with targeting moieties such as monoclonal antibodies or their fragments. At present, most of the effort in vaccine and API delivery is focused on LNPs. However, nanoparticles formed from biodegradable polymers such as polylactic-co-glycolic acid (PLGA) also offer an excellent alternative approach to achieving controlled release.
“In microfluidic systems, nanoparticles are generated at the interface of two flows, frequently a lipid – which forms the particle – dissolved in an organic solvent (such as ethanol) and a hydrophilic cargo or drug dissolved in water or an aqueous buffer.”
PRODUCTION CHALLENGES
Current nanoparticle production methods tend to either be batch based or use relatively large-scale, continuous flow equipment, such as membranes. Both methods have significant drawbacks; the particle size is not very consistent and the encapsulation efficiency – the amount of drug or other cargo that ends up in the particle – can be poor, resulting in the waste of expensive materials. The minimum sample size is also typically large, causing significant problems in early-stage work where the high cost and limited availability of specialised lipids and the API restrict the amount of material available.
Many experiments need to be performed to optimise the formulation, so it is important to use as little material as possible during investigative studies and process optimisation. Furthermore, batch-based methods often require revalidation as the batch scale increases, as there are significant changes to mass transfer and other characteristics as vessels become larger. Microfluidics is an excellent solution to these problems, allowing experimental work with small quantities of materials, and offering exceptional particle size monodispersity and encapsulation efficiency, and the ability to scale up to high throughput with no change to the fundamental particle formation method.
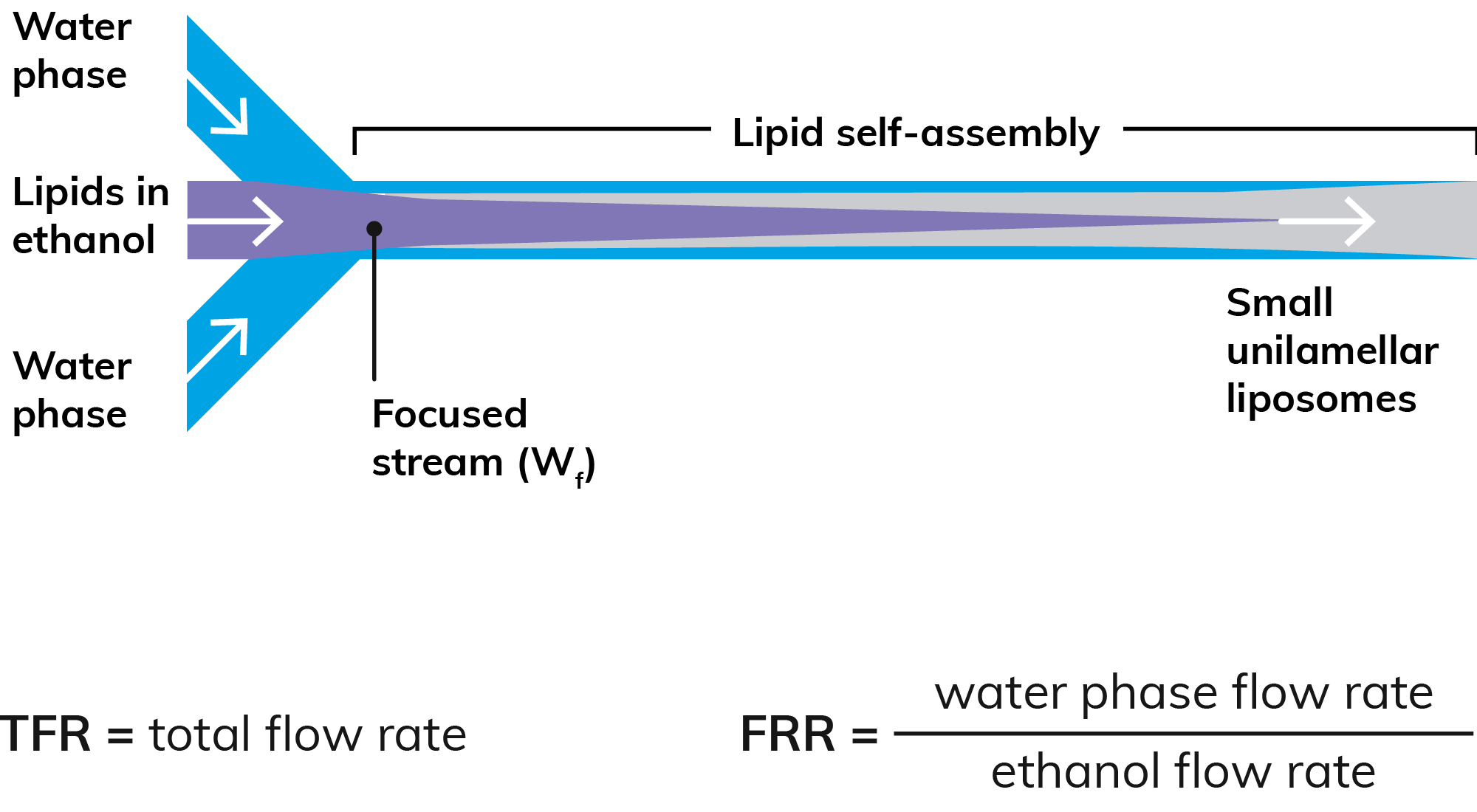
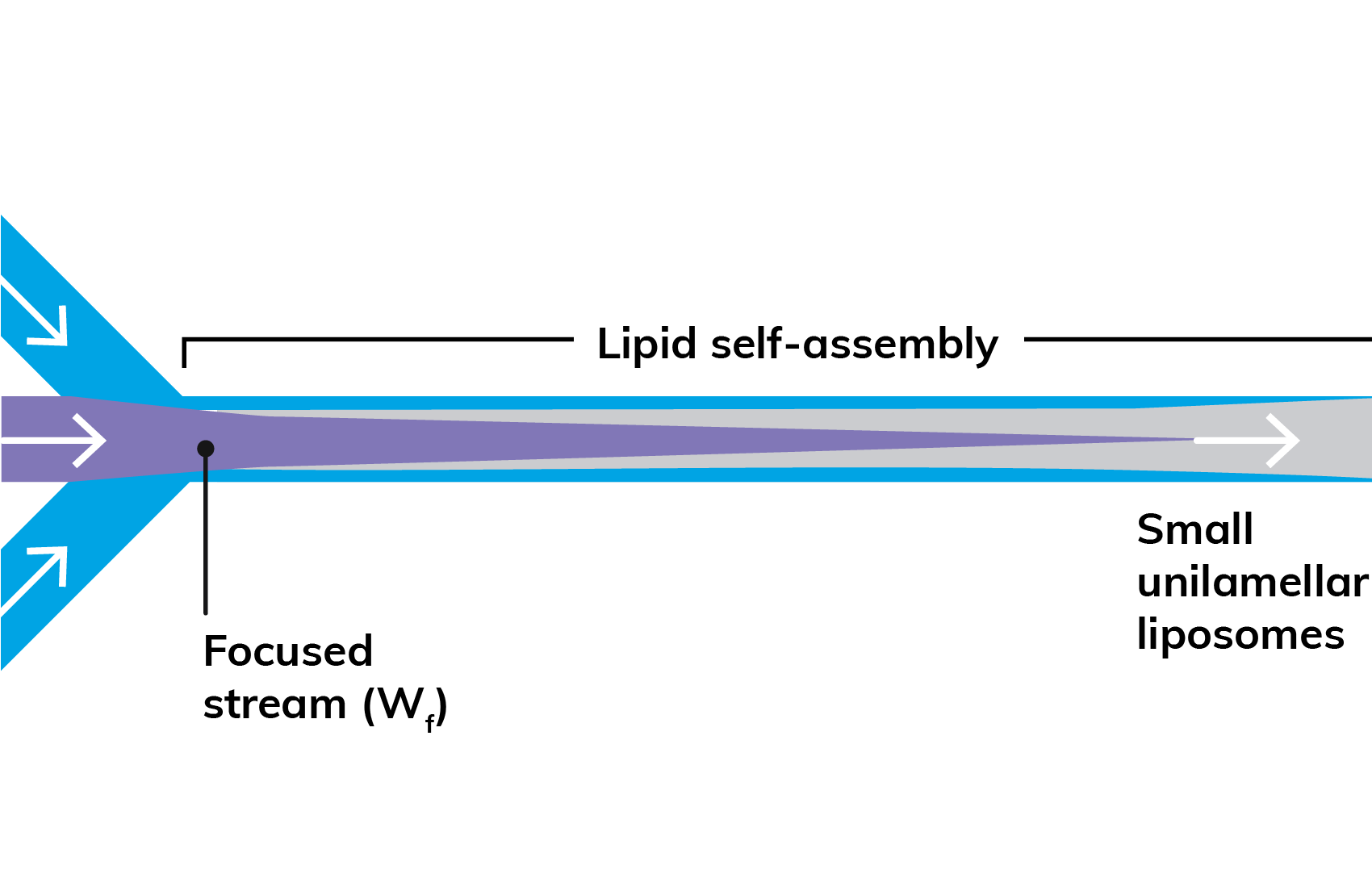
Figure 1: Microfluidic production of nanoparticles.
MICROFLUIDIC GENERATION OF NANOPARTICLES
In microfluidic systems, nanoparticles are generated at the interface of two flows, frequently a lipid – which forms the particle – dissolved in an organic solvent (such as ethanol) and a hydrophilic cargo or drug dissolved in water or an aqueous buffer. When these immiscible streams meet, the lipid precipitates, then self-assembles into spherical particles encapsulating the drug (Figure 1). The resulting nanoparticle size is typically a function of LNP composition and flow rate ratio (FRR), while the polydispersity index (PDI) and encapsulation efficiency can be optimised by careful choice of flow conditions and lipid/stabiliser selection. However, like batch methods, first-generation microfluidic tools are best suited to processing a single set of reagents, making it hard to vary production protocols quickly and automatically to generate test samples from a range of lipids, polymers and APIs.
A DEMAND FOR AUTOMATION
Clearly, there is an urgent need for automated production of LNP libraries for screening and optimisation, and subsequent scale-up for cGMP production. Automation would streamline screening of nanoparticles to allow the selection of candidates with the most promising biological performance. It would also enable rapid optimisation of particle size, consistency and encapsulation efficiency – reducing cost and increasing throughput. Equally importantly, it would also enable the scale-up of the development protocol to larger-scale manufacturing without redefining and re-optimising the process.
Typical performance requirements for automated LNP production include the ability to generate a wide range of particle sizes and to modify a variety of parameters quickly and easily – for example, to evaluate different flow rates or temperatures. Pharmaceutical companies often automatically produce a plethora of drug analogues, which are then screened using automated biological assays to determine which candidates to take forward. An automated LNP system would allow rapid screening of candidate formulations against a wide range of biological targets. It is, of course, vital that any automated nanoparticle generation system integrates seamlessly with the laboratory information management system to allow validated data capture and processing, and integration with upstream and downstream processes.
A NOVEL PRODUCTION PLATFORM
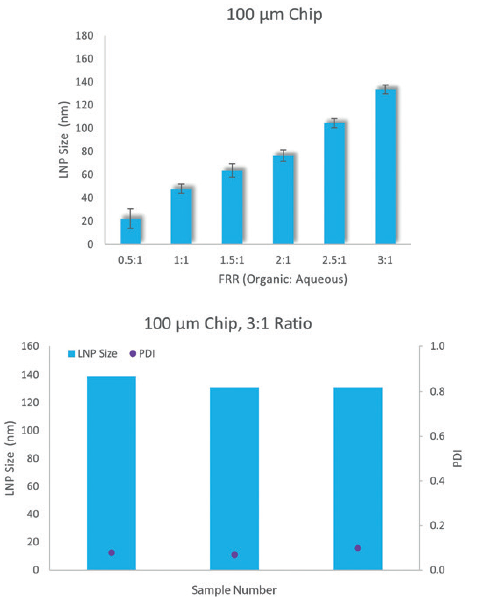
Figure 2: Particle size is consistent over a range of FRRs and relatively insensitive to the TFR for a given FRR.
The need to overcome the challenges of protocol development and enable subsequent scale-up to GMP manufacturing led one company specialising in the field of microfluidics to develop a dedicated nanoparticle production platform offering automated set-up and execution of a sequence of samples. Lipid and cargo are loaded into software-controlled reagent injection loops for serial injection of samples – the sole manual step in the protocol. A series of experiments is then executed, with pump operation, loop switching and washing performed automatically. The resulting nanoparticle samples are collected using an automated liquid handler. Particles ranging in size from 20 to 800 nm can be readily and repeatably generated, with an excellent PDI and encapsulation efficiency, for cost-effective use of expensive APIs for process development.
It is important that the LNP size and PDI are consistent from run to run. The use of 5 mL sample loops and ~250 μL samples allows multiple parameters to be explored in a single run, with automated washing of all wetted parts between samples to eliminate cross-contamination. Particle size has been shown to be consistent over FRRs ranging from 0.5:1 to 3.5:1, while being relatively insensitive to the total flow rate (TFR) (Figure 2).
Once the desired particle size is achieved, other parameters – such as PDI, stability and charge – may be improved by using different lipids, or combinations of lipids and/or stabilisers such as cholesterol, for improved nanoparticle stability. Solubility and viscosity affect the formation rate, and processes can be optimised with different reagents by careful selection of an appropriate microfluidic chip. Chips are available that work at a low TFR for efficient use of costly reagents and APIs, while others allow operation at a very high TFR for high-throughput particle generation. Production rates for the protocol development system range from 100 μL/min to 16 mL/min per channel, allowing work with small amounts of material in early stages and later production of trials samples at higher throughput. Small amounts of sample – from 200 μL to 5 mL – can be generated during the development phase then, once the protocol is established, the process can be scaled up by operating the same system in continuous production mode for larger-scale manufacture of up to 1 L/h, without the need for revalidation. This offers the benefit of fast and economical operation, with a single system able to perform all the necessary tasks.
LNP PRODUCT AND PROCESS DEVELOPMENT
Start With the End in Mind
This newly developed automated nanoparticle system is now the focus of a collaboration with a leading contract development and manufacturing organisation (CDMO), with the goal of establishing a robust platform for LNP and polymeric particles to support nanoparticle product development, validation, scale-up and subsequent GMP production. This collaboration will provide the foundation needed for the CDMO’s partners to advance their projects into clinical development in the most effective way.
A new project typically starts with a thorough analysis of product requirements from a clinical and commercial standpoint and goal setting. As a first step, the target product profile (TPP) has to be established. The TPP has emerged as an important tool that enables planning and communication between all key stakeholders, sponsors and regulatory authorities, and helps to establish the efficacy, safety and commercial suitability of the product. The US FDA issues guidance on the TPP for better communication between product developers and regulatory agencies.
The TPP for nanoparticles typically includes the following product characteristics:
- Dose – in the early stage it can be a range of doses that is further defined after dose-ranging clinical studies, typically in Phase II.
- Concentration of the final drug product (DP).
- Yields – the cost of the raw materials (both cargo and excipients: lipids, polymers) is very significant, so waste needs to be minimised.
- Stability – both chemical and colloidal stability need to be evaluated. Frozen product can be used for early-stage clinical trials to accelerate timelines but, for commercial products, a formulation stable at room temperature or 4 °C (refrigerated conditions) is much preferred.
- Container closure/presentation – single dose, multi-dose, vial and syringe or prefilled syringe presentation?
For multi-component systems, the process is critical, so it is very important to ensure control over the product properties, process robustness and reproducibility. A successful approach can best be described as “the product is the process”, as control of the product properties is best achieved through the control of the process and the associated unit operations.
Key unit operations and critical process parameters must be defined early in the development process and include LNP batch fabrication formulation and downstream operations (Figure 3; DP unit operations, such as aseptic fill-finish, and labelling/packaging, are not included). In addition, robust analytical methodology needs to be established to ensure control of the process.
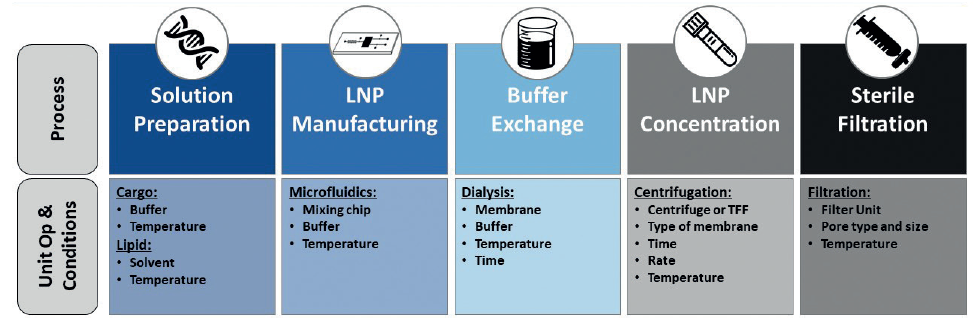
Figure 3: Key stages of LNP product and process development.
One of the key aspects of developing LNP-based formulations with the desired target properties is the composition. Typically, LNPs contain the following lipid components:
- Ionisable lipid, positively charged at low pH (enabling RNA complexation)
- Helper lipid that contributes to delivery efficiency and stability
- Structural lipid, such as cholesterol
- PEGylated lipid to minimise reticuloendothelial clearance and increase circulation time.
The development protocol and formulation of the LNP are achieved using design of experiments optimisation to balance efficacy and tolerability, and to ensure that the overall manufacturing processis as effective as possible. This includes establishing robust and reproducible processes that can be scaled up or down, along with LNP drug product development incorporating formulation, stability studies and container closure comparability at an early stage.
As previously mentioned, robust analytical methodology must be established early in the development process. As per April 2018 FDA Guidance for Industry “Liposome Drug Products” this should include the following key test methods:
- Particle size
- Particle size distribution, PDI
- Zeta potential
- mRNA identity and concentration
- mRNA encapsulation efficiency (%)
- Lipid identity and purity
- pH and osmolarity
- Residual solvent
- Sterility (endotoxin and bioburden)
- Stability: 4 °C and/or frozen suspension
- Content uniformity (mRNA in the vial).
To date, the automated nanoparticle system has been used successfully to fabricate mRNA LNPs with controlled particle size between 80 and 150 nm throughout the process – scaling up and down without having to redevelop the protocol – a modest size increase post dialysis/concentration and mRNA encapsulation above 90%.
THE WAY OF THE FUTURE
Building on the success to date, there are now plans to develop a fully automated, lab-scale walk-away system able to generate libraries of 10 to 96 samples per run from precursor lipids and cargo, using septum-sealed 96-well plates to prevent cross-contamination and avoid evaporative losses. Predefined user data files uploaded to the system software will allow easy input of preparation protocols, including lipid and cargo selection, flow rates, in-line dilutions and heart cut sample collection, as well as the volume to be collected. The platform will enable collection of samples ranging in volume from 250 μL to 2 mL in sealed well plates, with a full system wash of all wetted parts between samples to ensure there is no cross contamination.
Once validated, this system will form the basis of a future modular high-throughput cGMP system incorporating sterilisable multiple multichannel microfluidic chips – typically offering a 100-fold increase in production throughput, compared with a single channel – and the capability to operate in continuous flow at up to 250 mL/min per channel to allow production at rates from 1–15 L/h per module.
CONCLUSION
Automated LNP production offers a seamless complete pathway from protocol development to GMP manufacture, without the need to change and revalidate methods. Researchers now have a tool for more effective, flexible and cost-effective protocol development and optimisation, as well as library generation, with better control of particle size distribution and encapsulation efficiency. Crucially, the process is scalable, by operating in continuous flow and by incorporating multiple microfluidic chips working in parallel, ensuring that the same method can be used from development right through to GMP production.
The authors believe that the commercial model for this development work should be “no strings attached”. This means that the development work is fairly valued and paid for as it is performed, without a requirement for royalties after the product has launched. Royalty-based approaches add an additional barrier and commercial risk into the cost model, increasing costs for successful products and reducing the collaborative drive during the critical stages of the development process. Open access to the microfluidics platform and associated services would enable rapid and cost-effective development of the next generation of innovative nanoparticles products.
LNP development requires a strong collaborative effort between product developers and process engineers and the support of an experienced CDMO. It is vital that all key aspects of the product and process are evaluated, designed and built in early in the development cycle to avoid unnecessary development timeline delays.