
Citation: Bicker M, Müller M, Mittermüller M, Haines D, Rothhaar U, “Comparative Extractable Studies for Injectables and Medical Devices Aligned with USP <1663> and ISO 10993 Guidelines, ONdrugDelivery, Issue 120 (May 2021), pp 86–95.
Matthias Bicker, Michael Müller, Marc Mittermüller, Daniel Haines and Uwe Rothhaar discuss the regulatory requirements that need to be considered when designing an extractables and leachables study for a drug product or medical device. To illustrate the subject further, the authors provide two example studies, each following a different set of regulatory guidelines.
In order to conduct best practices for the safety assessment of materials used for pharmaceutical drug product packaging and medical devices, the most recent regulatory guidance needs to be considered. For chemical characterisation of components and materials, different regulatory guidelines focused on extractables have been established. United States Pharmacopeia (USP) <1663> provides a framework for an extractables assessment of pharmaceutical packaging and drug delivery systems.1 The principles of this framework are recommended for pharmaceutical development, manufacturing applications and medical device components related to combination products. USP <1663> comprises scientific principles and best practices recommended for the manufacturer of drug substances and drug products as well as manufacturers of pharmaceutical and medical device packaging.
“Secondary packaging components and components with indirect drug contact, such as labels, need to be taken into account as potential sources of chemical compounds that can migrate into the drug product.”
ISO 10993 addresses the evaluation of medical devices with respect to their biological safety. An important part of this framework is the new revision of ISO 10993-18,2 focused on chemical characterisation of medical device materials within a risk management process. The scope of this new revised guideline is the identification and quantification of the chemical constituents of medical devices in a stepwise approach, including an estimation of the potential of the medical device to release chemical substances (extractables) and a measurement of released chemical substances (leachables). The new ISO 10993-18 revision emphasises a greater integration and harmonisation with ISO 10993-1 (a general framework for planning of biological evaluation and testing within a risk management process), ISO 10993-12 (recommendations for sample preparation and specific extraction conditions) and ISO 10993-17 (allowable limits for leachable substances).3–5
Further frameworks have been established by the Product Quality Research Institute (PQRI)6,7 with general and specific recommendations for extractables and leachables (E&L), by the EMA with a guideline focused on “plastic immediate packaging materials” addressing the need for testing of the compatibility of the plastic material with the medicinal product by performing extraction studies,8 and by USP <661>9 among others. USP <661> is a further standard for plastics used to package medical devices, which will be substituted and expanded upon by USP <661.1> for plastic materials of construction and USP <661.2> for plastic packaging for pharmaceutical use. In addition to the implemented ICH Q3D standard for elemental impurities,10 a new chapter for organic impurities, “ICH Q3E: Assessment and Control of Extractables and Leachables for Pharmaceuticals and Biologics”, is currently under development.11 Different levels of identification have been suggested for extractables studies, including “partial”, “tentative”, “confident” and “confirmed”, with increasing certainty that the identification is correct.12
SCHOTT Pharma Services offers analytical services for extractables and leachables testing and related chemical characterisation of primary packaging and medical device components and materials.13,14 These services are aligned with the requirements presented by customers and the most recent regulatory guidelines. To address the demands of E&L characterisation, this article covers the importance of the following systematic steps for conducting an effective extractables study.
PROCEDURE – HOW TO GATHER THE RIGHT INFORMATION TO SET UP AN EXTRACTABLES STUDY
The overall procedure to collect the underlying information for extractable studies typically comprises some or all of the following steps:
- Clarification of the customer’s request, product application (drug or medical product and associated packaging or device) and related requirements supported by scientific consulting.
- Scientific advice concerning any relevant regulatory guidelines.
- Support for analytical evaluation threshold (AET) calculation based on ICH M7,15 USP <1663>,1 ISO 10993-182 or PQRI16 recommendations.
- Recommendation of appropriate study design for drug product application, including a detailed study protocol and suitable extraction method (e.g. sealed vessel extraction with shaking incubation, reflux or Soxhlet extraction).
- Extractables study, according to the recommendations from one or more of:
– USP <1663>,1 including exaggerated or simulated extraction conditions
– ICH Q3D10
– USP <232>17
– PQRI7,18
– ISO 10993-18 or 10993-12,2,4 including exhaustive extraction or simulated extraction conditions. - Simulated in-use study (e.g. leachables from processing components) following current ISO 10993-18 recommendations.2
- Accelerated leachables studies or bridging studies to fill the gap between extractables and leachables.
- Collaboration with a toxicologist for alignment of organic and inorganic target compounds for a subsequent leachables study.
The results of such an extractables study together with the toxicological assessment are the basis for the target list of substances to be considered in a subsequent leachables study.
COMPONENTS – WHAT NEEDS TO BE CHARACTERISED WITHIN AN EXTRACTABLES STUDY?
Extractables data need to be generated for all materials with direct or indirect drug contact, and should be separately generated for each individual component. Additionally, secondary packaging components and components with indirect drug contact, such as labels, need to be taken into account as potential sources of chemical compounds that can migrate into the drug product. Typical applications for extractables characterisation are primary packaging components made of polymer, glass or elastomer (rubber material), or secondary packaging materials, labels on polymer packaging and other manufacturing components used by the pharmaceutical industry.
Components that need to be considered include:
- Primary packaging components of container closure systems, such as:
– Polymer syringe: plunger (rubber stopper), tip cap
– Glass syringe: plunger (rubber stopper), needle shield
– Glass cartridge: plunger (rubber stopper), rubber cap
– Glass vial: rubber closure
– Coated primary packaging components, such as siliconised components. - Labels with glue and ink (particularly required for polymeric primary packaging).
- Secondary packaging materials, such as:
– Nest
– Tub
– Cover sheet
– Bag
– Tray
– Secondary packaging components of medical devices, for example the polymer adapter or plunger rod. - Manufacturing components, such as:
– Silicone tubing
– Filters
– Carboys.
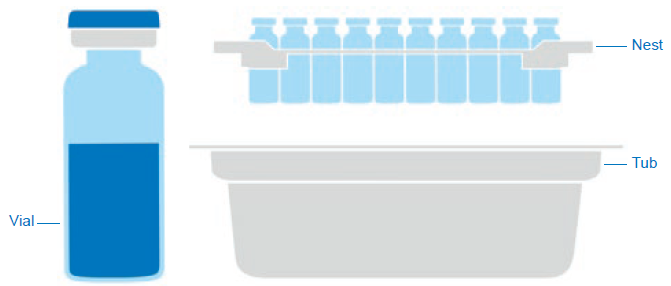
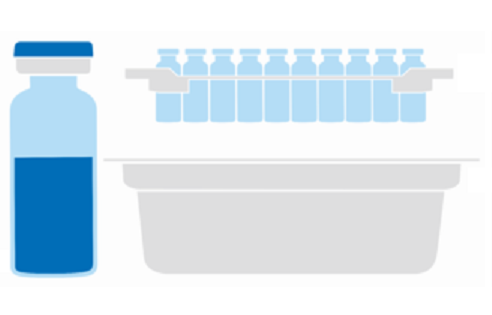
Figure 1: Components of a sterile ready-to-use packaging solution.
As an example, different components of a sterile nest/tub packaging solution for glass vials are shown in Figure 1.
The next section presents an example of a comparative extractable study for injectables and medical devices aligned with USP <1663> and ISO 10993 guidelines. The study is focused on a polymer syringe system consisting of a polymer barrel, polymer tip cap and elastomeric plunger (Figure 2). An example with polymer and elastomer components was chosen for this comparison because specific extraction conditions were recommended in both guidelines that should be applied for these materials.
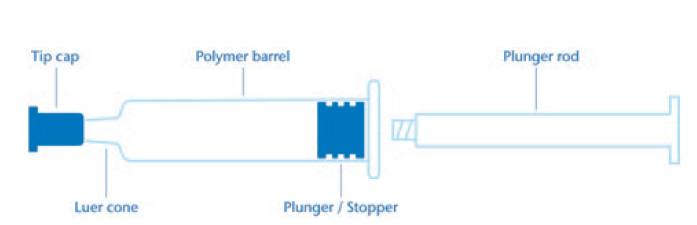
Figure 2: Polymer syringe system.
STUDY DESIGN – A SUITABLE ANALYTICAL PROCEDURE FOR AN EXTRACTABLES STUDY
The design used for an extractables study should be appropriate to identify organic and inorganic substances that are extracted when the components of a packaging system are exposed to suitable solvents, which are recommended by the regulatory guidance. The analytical methods used are:
- Gas chromatography – mass spectrometry (GC-MS)
– Used for determination and screening of semi-volatile organic compounds (SVOCs)
– Allows for the identification and quantification of low to medium molecular weight compounds, such as additives, catalysts, residual monomers and oligomers of polymers and rubbers, as well as semi-volatile plasticisers and processing agents. - Headspace gas chromatography – mass spectrometry (HS-GC-MS)
– Used for determination and screening of volatile organic compounds (VOCs)
– Allows for the identification and quantification of low molecular weight leachables, such as residual monomers of polymers or elastomers, residual solvents19,20 from component manufacturing and volatile oxidation and degradation products. - Liquid chromatography – mass spectrometry (LC-MS) and ultraviolet detection (LC-UV)
– Used for determination and screening of extractable and leachable non-volatile organic compounds (NVOCs)
– Allows for the identification and quantification of organic compounds with high polarity and medium to high molecular weight compounds, such as antioxidants (Figure 3), fatty acids from polymer and rubber component manufacturing and non-volatile plasticisers and processing agents. - High-resolution inductive coupled plasma mass spectrometry (HR-ICP-MS)
– Used to quantify the amounts of extractable and leachable elemental impurities
– Allows for the identification and qualification of elements of ICH Q3D classes 1–3 (summarised in Table 6).10 - Ion chromatography (IC)
– Used to quantify the amounts of extractable and leachable target anions. - Gravimetric non-volatile residue (NVR) analysis
– Used to determine the amount of non-volatile residue of solvent solution after extraction
– Allows for an assessment concerning the maximum total amount of non-volatile extractables and whether the extraction was exhaustive according to ISO 10993-12.4
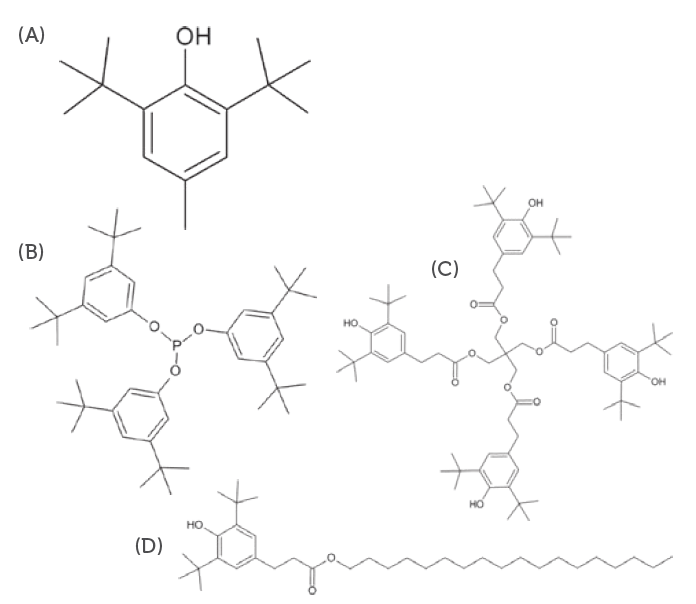
Figure 3: Typical antioxidants used as polymer addatives. (A) BHT, (B) Irgafos 168, (C) Irganox 1010 and (D) Irganox 1076.
Two examples of appropriate study designs for extractables studies conducted for polymer syringe components are illustrated in Table 1 and Table 2. The study design shown in Table 1 is aligned with current USP <1663> recommendations1 and the one in Table 2 is aligned with current ISO 10993-18 and 10993-12 recommendations.2,4
| Analytical method |
Neat material (*) |
Solvent | ||
| IPA: Water (50:50) |
Water (pH 5.2) |
Water (pH 9.5) |
||
| Headspace GC-MS (VOCs) |
X | – | – | – |
| GC-MS (SVOCs) | – | X | X | X |
| LC-MS and/or LC-UV (NVOCs) |
– | X | X | X |
| HR-ICP-MS (elemental impurities) |
– | – | X | – |
| IC (anions) | – | – | – | X |
Table 1: Tests to be performed during study design A (aligned with USP <1663>). (*) – direct analysis of neat sample material during thermal extraction.
| Analytical method |
Neat material (*) |
Solvent | ||
| n-Hexane | IPA | Ultrapure Water | ||
| Headspace GC-MS (VOCs) |
X | – | – | – |
| NVR | – | X | X | X |
| GC-MS (SVOCs) | – | X | X | X |
| LC-MS and/or LC-UV (NVOCs) |
– | X | X | X |
| HR-ICP-MS (elemental impurities) |
– | – | – | X |
| IC (anions) | – | – | – | X |
Table 2: Tests to be performed during study design B (aligned with ISO 10933). (*) – direct analysis of neat sample material during thermal extraction.
The studies also each use different extraction techniques and methods. Study protocol A is based on a reflux extraction technique, while study protocol B is based on a sealed vessel extraction technique and an exhaustive extraction method. Results from a practical example of this study are shown in Box 1.
BOX 1: TEST RESULTS OF EXTRACTABLES STUDIES
set of extractables studies for a commercial polymer syringe system were conducted using study protocols A and B. The syringe system under investigation comprises of a barrel made of polymer material and halobutyl rubber components (plunger, tip cap). Some selected results for antioxidants (polymer additives), non-volatile residues and concentrations of elemental impurities and anions in the following section are shown here (note: the peaks labelled with “IS” in the chromatograms belong to internal standard reference material for analysis – they were deliberately added and are not a syringe constituent).
HS-GC-MS
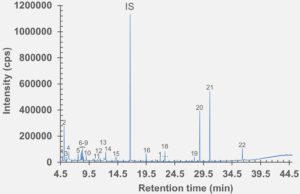
Figure 4: HS-GC-MS chromatogram of polymer syringe barrel component after incubation at 150 °C for 45 minutes.
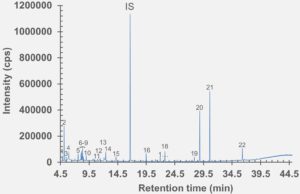
Figure 5: HS-GC-MS chromatogram of rubber stopper component after incubation at 150 °C for 45 minutes.
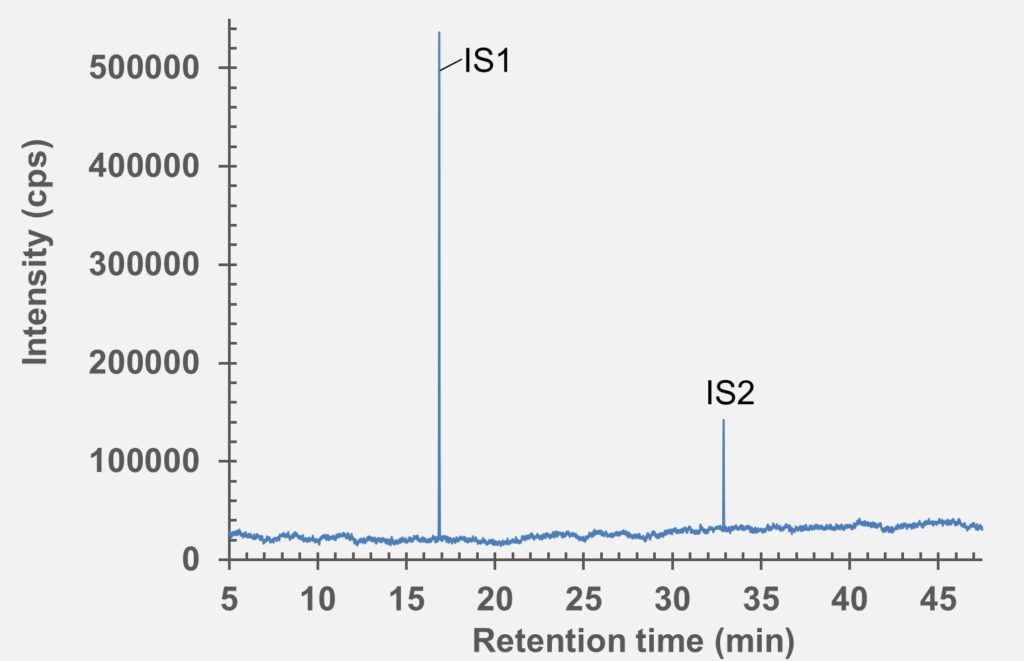
Figure 6: GC-MS chromatogram of polymer syringe barrel after extraction in water (pH 5.2).
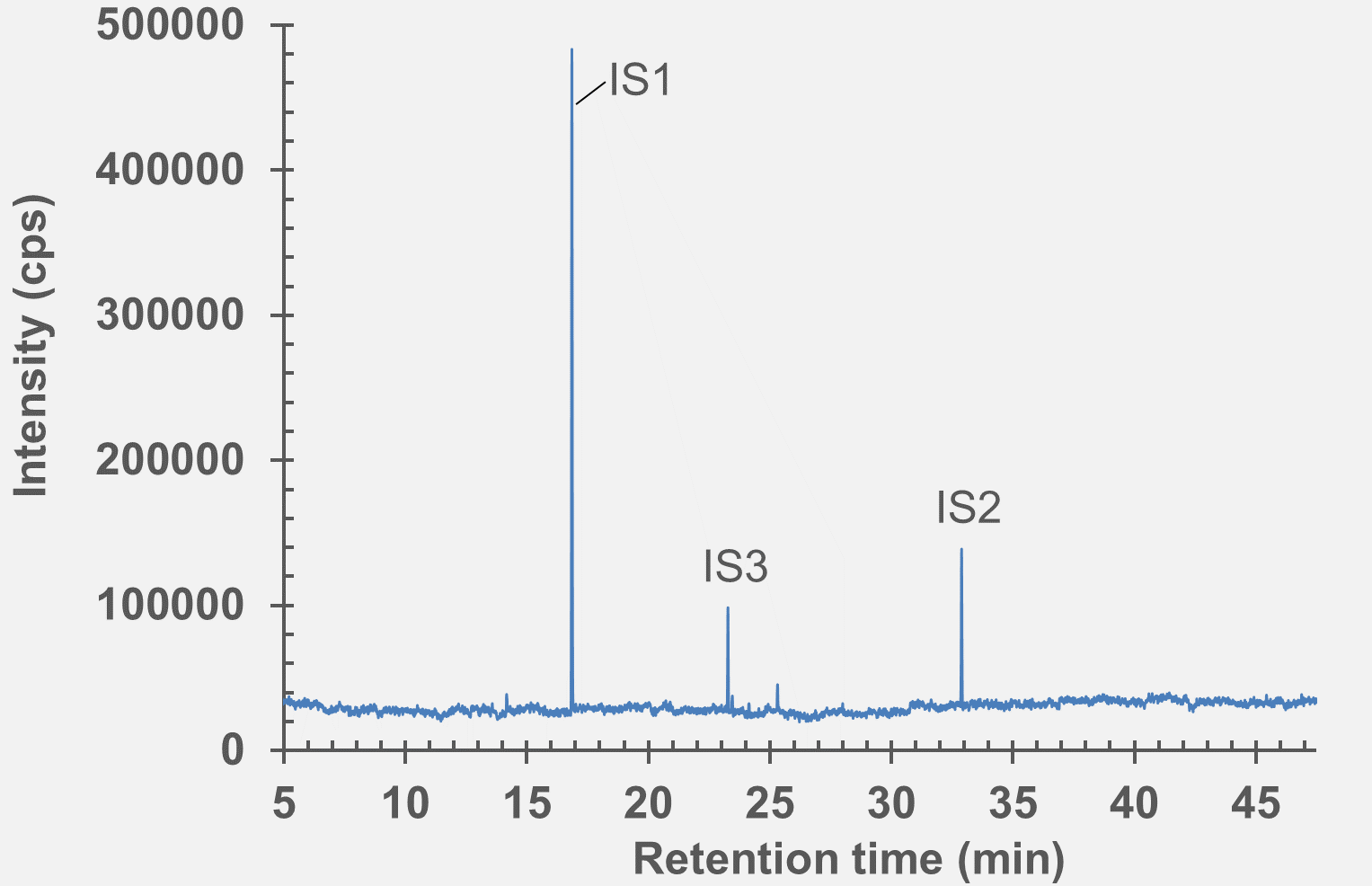
Figure 7: GC-MS chromatogram of polymer syringe barrel after extraction in water (pH 9.5).
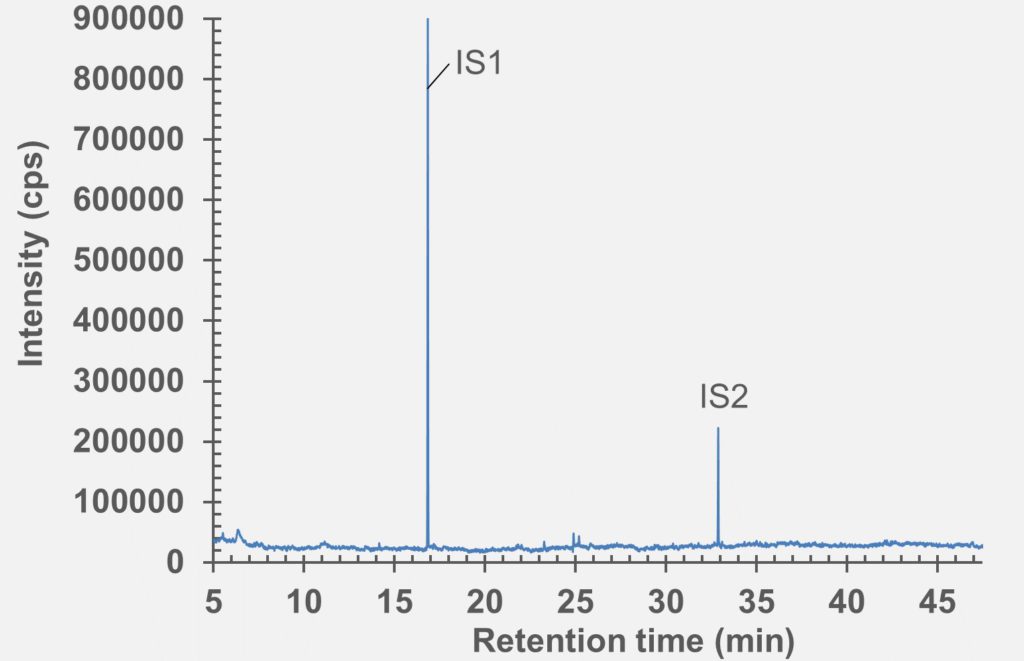
Figure 8: GC-MS Chromatogram of polymer syringe barrel after extraction in IPA/water (1:1).
LC-MS and LC-UV
| Antioxidant / degradation product |
Polymer syringe barrel (Irradiation sterilised) |
Halobutyl rubber tip cap (irradiation sterilised) |
Halobutyl rubber stopper (irradiation sterilised) |
||||||
| Water pH 5.2 |
Water pH 9.5 |
IPA: Water (50:50) |
Water pH 5.2 |
Water pH 9.5 |
IPA: Water (50:50) |
Water pH 5.2 |
Water pH 9.5 |
IPA: Water (50:50) |
|
| BHT | < RL | < RL | < RL | < RL | < RL | 5.4 | < RL | < RL | 5.7 |
| BHT aldehyde | < RL | < RL | < RL | < RL | < RL | < RL | < RL | 0.08 | 0.35 |
| Irganox 1010 | < RL | < RL | < RL | < RL | < RL | < RL | < RL | < RL | < RL |
| Irganox 1076 | < RL | < RL | < RL | < RL | < RL | < RL | < RL | < RL | < RL |
| Irgafos 168 | < RL | < RL | < RL | < RL | < RL | < RL | < RL | < RL | < RL |
| Irgafos 168 oxidised | < RL | < RL | < RL | < RL | < RL | < RL | < RL | < RL | < RL |
| All amounts in (μg/unit) | |||||||||
Table 3: LC-MS results of quantities of antioxidants found in components of a prefillable syringe system after extraction with aqueous and mixed solvents aligned with USP <1663>.1 Reporting limit (RL) was within the range 0.05–1.0 μg/unit.
| Antioxidant / degradation product |
Polymer syringe barrel (Irradiation sterilised) |
Halobutyl rubber tip cap (irradiation sterilised) |
Halobutyl rubber stopper (irradiation sterilised) |
||||||
| Ultra pure water |
IPA | Hexane | Ultra pure water |
IPA | Hexane | Ultra pure water |
IPA | Hexane | |
| BHT | < RL | < RL | < RL | < RL | 14 | 47 | < RL | 19 | 19 |
| BHT aldehyde | < RL | < RL | 19 | 0.06 | 0.49 | 1.5 | 0.06 | 0.67 | 1.2 |
| Irganox 1010 | < RL | < RL | 393 | < RL | < RL | 0.22 | < RL | < RL | < RL |
| Irganox 1076 | < RL | < RL | < RL | < RL | < RL | 0.09 | < RL | < RL | 0.04 |
| Irgafos 168 | < RL | < RL | < RL | < RL | < RL | < RL | < RL | < RL | < RL |
| Irgafos 168 oxidised | < RL | < RL | < RL | < RL | < RL | 0.34 | < RL | < RL | 0.08 |
| All amounts in (μg/unit) | |||||||||
Table 4: LC-MS results of amounts of antioxidants found in components of a prefillable syringe system after extraction with polar and non-polar solvents according to ISO 10993-18 and 10993-12. Reporting limit (RL) was within the range 0.02–1.2 μg/unit.
NVR
| Extraction cycle no. | Polymer syringe barrel (Irradiation sterilised) |
Halobutyl rubber tip cap (irradiation sterilised) |
Halobutyl rubber stopper (irradiation sterilised) |
|||
| IPA | Hexane | IPA | Hexane | IPA | Hexane | |
| 1 | < 0.35 | 16 | 1.2 | 20 | 0.7 | 8.0 |
| 2 | < 0.35 | 8.4 | 0.7 | 4.4 | 0.4 | 1.1 |
| 3 | – | 5.2 | 0.4 | 1.2 | 0.2 | 0.3 |
| 4 | – | 4.0 | 0.2 | – | 0.2 | – |
| All amounts in (μg/unit) | ||||||
Table 5: Results of gravimetric determination of NVR shown for up to four extraction loops. Extraction was conducted at 50 °C for 72 hours, in accordance with ISO 10993-12.4
HR-IDP-MS
| Classification ICH Q3D |
Elements Tested | Classification ICH Q3D |
Elements Tested |
| Class 1 | As, Cd, Hg, Pb | Class 4 | Ba, Cr, Cu, Li, Mo, Sb, Sn |
| Class 2 | Co, Ni, V | Other elements | Al, B, Ca, Fe, K, Mg, Mn, Na, W, Zn |
| Class 3 | Ag, Au, Ir, Os, Pd, Pt, Rh, Ru, Se, Tl | Additional | Si, Bi, Ce, Hf, P, S, Ti, Zr |
Table 6: Overview of elemental impurities listed in ICH Q3D and USP <232> by their classification10,17 and additional elements tested for.
| Elements | Classification according to ICH Q3D |
Polymer syringe barrel (Irradiation sterilised) |
Halobutyl rubber tip cap (irradiation sterilised) |
Halobutyl rubber stopper (irradiation sterilised) |
|||
| Water pH 5.2 |
Ultrapure water |
Water pH 5.2 |
Ultrapure water |
Water pH 5.2 |
Ultrapure water |
||
| As, Cd, Hg, Pb | class 1 | < RL for all class 1 elements for all extracts of all components | |||||
| Co, Ni, V | class 2A | < RL for all class 2A elements for all extracts of all components | |||||
| Ag, Au, Ir, Os, Pd, Pt, Rh, Ru, Se, Tl | class 2B | < RL for all class 2B elements for all extracts of all components | |||||
| Ba, Cr, Cu, Li, Mo, Sb, Sn |
class 3 | < RL for all class 3 elements for all extracts of all components | |||||
| Ca | – | < RL | < RL | 1.4 | 1.5 | 0.226 | 0.41 |
| Mg | elements | < RL | < RL | 18 | 17 | 2.6 | 2.1 |
| Al, B, Fe, K, Mn, Na, W, Zn | < RL for respective “other elements“ for all extracts of all components | ||||||
| Si | Additional (not classified in ICH Q3D) | < RL | < RL | < RL | 6.6 | < RL | < RL |
| Bi, Ce, Hf, P, S, Ti, Zr |
< RL for respective additional elements for all extracts of all components | ||||||
| All amounts in (μg/unit) | |||||||
Table 7: Results of HR ICP-MS analyses for elemental impurities after extraction in water (pH 5.2) or in ultrapure water.
Reporting limit (RL) for class 1–3 elements in the range of 0.003–0.33 μg/unit, for “other elements” of ICH Q3D in the range of 0.063–12 μg/unit and for additional elements in the range of 0.003–25 μg/unit.
IC
| Extraction cycle no. |
Polymer syringe barrel (Irradiation sterilised) |
Halobutyl rubber tip cap (irradiation sterilised) |
Halobutyl rubber stopper (irradiation sterilised) |
|||
| Water pH 9.5 | Ultrapure water | Water pH 9.5 | Ultrapure water | Water pH 9.5 | Ultrapure water | |
| Acetate (CH3COO-) | < RL | < RL | 2.8 | 4.1 | < RL | < RL |
| Bromide (Br-) | < RL | < RL | 6.5 | 6.2 | 2.5 | 2.6 |
| Chloride (Cl-) | < RL | < RL | < RL | 0.43 | < RL | < RL |
| Fluoride (F-) | < RL | < RL | < RL | < RL | < RL | < RL |
| Formate (HCOO-) | < RL | 2.3 | 32 | 34 | < RL | 0.47 |
| Nitrate (NO3-) | < RL | < RL | 3.8 | 3.5 | 0.78 | < RL |
| Phosphate (PO43-) | < RL | < RL | < RL | < RL | < RL | < RL |
| Sulfate (SO42-) | < RL | < RL | < RL | < RL | < RL | < RL |
| All amounts in (μg/unit) | ||||||
Table 8: Results of IC analyses for target anions after extraction in water (pH 9.5) or in ultrapure water.
“For material and extractables characterisation of polymeric components, all guidelines have many aspects in common. However, as can be seen by the approaches taken by study designs A and B, the guidelines partially differ in details of the procedures and extraction conditions they recommended.”
Analysis of VOCs by HS-GC-MS
- Incubation of neat sample material at 150°C for 45 minutes in sealed vessels
- Qualitative and semi-quantitative screening analysis of an aliquot of the respective gas phases for VOCs by HS-GC-MS
- Identification and semi-quantitative evaluation of substance signals using commercial and internal databases and suitable internal standards.
Extraction of SVOCs and NVOCs
Study protocol A:
- Reflux extraction of samples in 50:50 isopropyl alcohol (IPA) and water, water (pH 5.2) and water (pH 9.5).
Study protocol B:
- Exhaustive extraction conditions based on determination of NVR:
– Evaporation of extract solutions from each extraction cycle to dryness for aliquots of each extract
– Determination of NVR by gravimetric analysis of dry residue
– Pooling of extracts from relevant extraction cycles for subsequent GC and LC analyses. - Exhaustive extraction of samples in ultrapure water, IPA and n-hexane
- Multiple extraction cycles (incubation condition: 50°C, 72 hours, under agitation) performed, depending on the results of individual NVR determinations.
Analysis of SVOCs by GC-MS
- Liquid-liquid extraction of aqueous extracts with dichloromethane (DCM) at different pH values and subsequent pooling of organic phases, followed by concentration of extracts if necessary
- Qualitative and semi-quantitative screening analysis of prepared extracts SVOCs by GC-MS
- Identification and semi-quantitative evaluation of substance signals by using commercial and internal MS databases and suitable internal standards.
Analysis of NVOCs by LC-MS and/or LC-UV
- Liquid-liquid extraction of aqueous extracts with dichloromethane (DCM) at different pH values and subsequent pooling of organic phases, followed by concentration of extracts and reconstitution in isopropyl alcohol if necessary
- Target screening for typical polymer additives, and qualitative and semi-quantitative screening analysis of prepared extracts for NVOCs by LC-MS and/or LC-UV
- Identification and semi-quantitative evaluation of substance signals by using high-resolution, time-of-flight MS, internal databases and suitable internal standards.
Extraction of Inorganic Elemental Impurities and Anions
Study protocol A:
- Samples for HR-ICP-MS analysis reflux extracted in water (pH 5.2)
- Samples for IC analysis reflux extracted in water (pH 9.5).
Study protocol B:
- Pooling of extracts from relevant extraction cycles using ultrapure water for both HR-ICP-MS and IC analysis.
Analysis of Inorganic Elemental Impurities and Anions
- Analyses of resulting extracts by HR-ICP-MS for elemental impurities and reporting of up to 40 elemental impurities including all class 1–3 elements outlined in ICH Q3D and USP <232> guidelines.10,17
- Analyses of resulting extracts by IC for the following target anions: acetate, formate, bromide, chloride, fluoride, nitrate, phosphate and sulfate.
RATIONALE BEHIND THE STUDY PROTOCOLS
Study Design A – USP <1663>
Study design A and the respective extraction conditions are aligned with the USP <1663> guideline for “assessment of extractables associated with pharmaceutical packaging/delivery systems”,1 where the general framework of scientific principles and best practices for extractables studies is described. According to USP <1663>, extractable studies are required due to the potential exposure of patients to leachable substances that could migrate from the pharmaceutical packaging or delivery system into the drug product, therefor it is important to assess the safety risk to the patient and any other potential issues posed by leachables.
For a leachables assessment, it is required to “know the identities and the levels to which leachables will accumulate in the finished drug product over its shelf life”. Since the primary and secondary packaging components are the “primary sources of potential leachables”, performing an extractables study on these components is justified.
Depending on the chemical nature of the drug formulation and its route of administration, specific examples are given in USP <1663>. For a small-volume parenteral drug product application based on an aqueous formulation (e.g. a drug product dissolved in a formulation with a pH value of 6.5), extractions of rubber components with three different solvents are recommended “to reflect the chemical nature of the formulation”:
- Aqueous acidic (pH 5.2)
- Aqueous alkaline (pH 9.5)
- Mixed aqueous and organic – IPA and water (50:50).
Study design A has been shown to be well suited for following all relevant USP <1663> guideline recommendations in numerous studies SCHOTT has conducted for its customers.
Study Design B – ISO 10993-18 and ISO 10993-12
Study design B is primarily based on the recommendations of the new ISO 10993-18 guideline, which outlines a chemical characterisation of medical device materials within a risk management process. A framework is specified in this guideline for “the identification and, if necessary, quantification of constituents of a medical device, allowing the identification of biological hazards and the estimation and control of biological risks from material constituents”.2
For extractable studies, ISO 10993-18 is focused on the chemical characterisation procedure. Related to extraction conditions, ISO 10993-18 integrates and refers to ISO 10993-12.4 For chemical characterisation of polymer components, an exhaustive extraction concept is recommended in ISO 10993-12. According to this guideline, the quantities of low molecular weight compounds (LMWCs) of polymers, such as additives, catalysts, residual monomers and oligomers, that can potentially migrate into the drug or medical product (and subsequently into the human body) should be determined, and an exhaustive extraction using both polar and non-polar solvents should be applied.
An extraction is defined as exhaustive if the residues extracted by a subsequent extraction are below 10% of the amounts found after the first extraction. In the case of polymer components used in medical devices, it needs to be confirmed within the extractables study that the extraction was exhaustive. For this purpose, a gravimetric method is recommended. In study design B, the NVR is determined using a gravimetric method for confirmation that the extraction was exhaustive. Study protocol B has been established to fulfil the requirements of both ISO 10993-18 and ISO 10993-12.
“It is a mandatory precondition for an appropriate extractables study design to review the drug product or medical device application and the route of administration in detail in order to align with the relevant regulatory requirements, which can vary by region, and to obtain a common understanding of the purpose of the study.”
DIFFERENCES BETWEEN ISO AND USP GUIDANCE AND PQRI RECOMMENDATIONS
For material and extractables characterisation of polymeric components, all guidelines have many aspects in common. However, as can be seen by the approaches taken by study designs A and B, the guidelines partially differ in details of the procedures and extraction conditions they recommended.
USP <1663> has a special focus on the chemical properties of the drug formulation (e.g. “many drug products are compositionally intermediate between polar and non-polar”) and on the route of administration. For example, for simulation of a worst-case leachable profile, solvents should be applied for extraction that have a similar or greater propensity for extraction of substances than the drug formulation.1
As another example, according to ISO 10993-12, the quantities of LMWCs of polymers should be determined based on an exhaustive extraction method applied with both polar and non-polar solvents.4 The strong focus on this exhaustive extraction method for polymer medical device components is only found in this guideline.
A hint for different concepts and recommendations depending on the applied regulatory guideline is also briefly mentioned in the new ISO 10993-18 guideline,2 including a short statement that exaggerated conditions might be requested by some legal authorities like the US FDA as credible alternatives to the ISO recommendation of exhaustive extraction. For medical device applications, this topic is also addressed by the FDA,22 which states that “a chemical analysis of the materials used in a device in its final finished form can be informative”, “can be used to assess the toxicological risk of the chemicals that elute from devices” and “chemical analysis using techniques per ISO 10993-12 can also be helpful to evaluate long-exhaustive extraction term toxicity endpoints, such as potential carcinogens”. On the other hand “chemical analysis is usually insufficient to identify all of the risks of the device in its final finished form, because it will not consider aspects of the finished device such as surface properties or device geometry that could affect the biological response in certain scenarios.” Furthermore, “extraction solvents should be selected to optimise compatibility with the device materials and provide information on the types of chemicals that are likely to be extracted in clinical use.”
As shown by the results data for LC-MS (Tables 3 and 4), the two study designs found significantly differing quantities of antioxidants (polymer and elastomer additives), which can be explained by the different extraction conditions and solvents. It is plausible that higher quantities of antioxidants were found in the non-polar hexane extracts, whereas very low quantities, sometimes even below the reporting limit, were found in the aqueous extracts due to their higher polarities. Nevertheless, the main purpose of an extractables study is the identification of potential leachables and both study designs met this goal, based on their respective regulatory frameworks.
Furthermore, the data for the HR-ICP-MS (Table 7) analyses showed comparable amounts of Ca and Mg found for the rubber components, even though aqueous solvents with different pH values and extraction conditions were used. Also, the data for IC analyses (Table 8) indicated that, for most anions, the extractable quantities were more or less comparable. These results indicate that similar quantities of the cations and anions were released for the aqueous solvents with different pH values and extraction conditions.
As a further example, according to PQRI recommendations for parenteral and ophthalmic drug products, extractables studies should be conducted with aqueous solvent solutions covering a very broad pH range between 2.5 and 9.5.18 This is based on the rationale that most aqueous drug product applications will be covered within this broad pH range because only a few therapeutic products have pH values outside of this pH range.
As a consequence, it is a mandatory precondition for an appropriate extractables study design to review the drug product or medical device application and the route of administration in detail in order to align with the relevant regulatory requirements, which can vary by region, and to obtain a common understanding of the purpose of the study. The identification of potential leachables within a customised extractables study can provide the basis for a subsequent toxicological assessment. Based on the toxicological assessment of the extractable profiles, the target compounds for the subsequent leachables study can be specified.
For leachables studies, method development and validation is required before determination of leachables in order to meet the required AET.
Relevant regulatory guidelines for leachable studies are:
- USP <1664>21
- USP <232>17
- USP <233>23
- ISO 10993-182
- ISO 10993-17.5
CONCLUSION
A deep understanding of the most recent regulatory guidelines, in particular the USP <1663> guideline and the new ISO 10993-18 international standard,1,2 is very important to give drug product-specific recommendations for an appropriate study design for extractables studies for pharmaceutical packaging, drug delivery systems and medical devices. Two different appropriate study designs, each primarily focused on one regulatory standard, and the applied analytical methods, have been described in detail, and example study results have been shown (Box 1) and the differences between them have been discussed.
SCHOTT Pharma Services provides analytical services, including extractables testing related to chemical characterisation of primary packaging and medical device components and materials. Such studies are designed with both customer requirements and the most recent regulatory guidelines in mind. Furthermore, SCHOTT provides leachables testing, including method development and method validation, following ICH Q2 (R1) recommendations24 and leachables characterisation based on USP <1664>, USP <232>, USP <233>, ICH Q3D and/or ISO 10993-18 and 10993-17 guideline recommendations.
SCHOTT Pharma Services’ laboratories are ISO/IEC 17025 accredited and FDA registered. SCHOTT Pharma Services has more than 40 years’ experience in analytical testing of pharmaceutical packaging containers. All quality relevant documents are electronically available, ensuring a hassle-free audit process.
REFERENCES
- “USP<1663> Assessment of extractables associated with pharmaceutical packaging/delivery systems”. US Pharmacopeial Convention, 2020.
- “ISO 10993-18: Biological evaluation of medical devices – Part 18: Chemical characterisation of medical device materials within a risk management process”. ISO, published 2020.
- “ISO 10993-1: Biological evaluation of medical devices – Part 1: Evaluation and testing within a risk management process”. ISO, published 2020.
- “ISO 10993-12: Biological evaluation of medical devices – Part 12: Sample preparation and reference materials”. ISO, published 2021.
- “ISO 10993-17: Biological evaluation of medical devices — Part 17: Establishment of allowable limits for leachable substances”. ISO, published 2002.
- Paskiet D et al, “Thresholds and Best Practices for Extractable and Leachables”. 3rd PQRI/FDA Conference on Advancing Product Quality, Washington DC, March 2017.
- Paskiet D et al, “The Product Quality Research Institute (PQRI) Leachables and Extractables Working Group Initiatives for Parenteral and Ophthalmic Drug Product (PODP)”. PDA J Pharm Sci Technol, 2013, Vol 67(5), pp 430–447.
- “Guideline on plastic immediate packaging materials”. EMA, published 2005.
- “USP<661> Plastic packaging systems and their materials of construction”. US Pharmacopeial Convention, 2020.
- “ICH guideline Q3D (R1) on elemental impurities”. EMA, published 2019.
- “ICH Q3E: Guideline for Extractables and Leachables (E&L)”. ICH, published 2020.
- Jenke D, “Identification and Quantitation Classifications for Extractables and Leachables”. PDA J Pharm Sci Technol, 2020, Vol 74(2), pp 275–285.
- Sögding T et al, “How Sterilisation of Primary Packaging Influences the Results of E&L Studies”. Contract Pharma, June 2015.
- “Pharma Analytics”. SCHOTT Pharma Services, web page.
- “ICH M7 Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk”. EMA, published 2017.
- Ball DJ, “Derivation of the Parenteral Safety Concern Threshold (SCT)”. PQRI PODP Extractables & Leachables Workshop, Apr 2018.
- “USP<232> Elemental Impurities – Limits”. US Pharmacopeial Convention, 2020.
- “Experimental Protocol for Qualitative Controlled Extraction Studies on Material Test Articles Representative of Prefilled Syringe (PFS) and Small Volume Parenteral (SVP) Container Closure Systems”. PQRI, Published 2011.
- “USP<467> residual solvents”. US Pharmacopeial Convention, 2020.
- “ICH Q3C (R6) Residual solvents”. EMA, published 2019.
- “USP<1664> Assessment of drug product leachables associated with pharmaceutical packaging”. US Pharmacopeial Convention, 2020.
- “Guidance for Industry: Use of International Standard ISO 10993-1 – Biological evaluation of medical devices – Part 1: Evaluation and testing within a risk management process”. US FDA, Sept 2020.
- “USP<233> Elemental Impurities – Procedures”. US Pharmacopeial Convention, 2018.
- “ICH Q2 (R1) Validation of analytical procedures: text and methodology”. EMA, published 1996.
