


To Issue 188
Citation: Hurlstone C, “Industrialising Drug Delivery Devices: Insights from 40 Years of Getting to Market”, ONdrugDelivery, Issue 188 (Jul 2026), pp 42–46.
Chris Hurlstone looks at the process of developing drug delivery systems through the industrialisation phase and explores ways to address the challenges that arise along the way. Drawing on Team’s 40-year track record of supporting drug delivery device innovation, the insights are intended for teams involved in developing drug delivery systems, particularly those responsible for design, engineering, industrialisation and manufacturing transfer.
Tools, technologies and processes may have changed over the decades, but many of the fundamentals of developing a drug delivery system (DDS) and taking it through the critical industrialisation phase remain unchanged. A DDS is defined as a given combination of a medical device and a medicinal product – whether provided as separate products or integrated as a single product – intended to deliver the medicinal product.
During DDS development, a lot depends on establishing the foundations for success from the outset. This involves ensuring a clear and viable product strategy, capturing requirements effectively and realistically, choosing concepts grounded in core engineering and scientific principles, and focusing strongly on design for manufacture and assembly from the word go.
UNDERSTAND AND BE GUIDED BY THE REGULATORY PATHWAY
Classifying a device or combination product and establishing its required regulatory pathway can often be straightforward. There are occasions however, particularly for innovative devices and technologies, where ambiguities arise, sometimes with multiple possible pathways to consider. Examples include whether or not to file as a substitutable generic, how to demonstrate bioequivalence or whether the device software is classified as Software as a Medical Device.
“THE DRUG DELIVERY DEVICE REGULATORY FRAMEWORK IS CONSTANTLY CHANGING AND THE LONG-RUNNING NATURE OF DEVICE DEVELOPMENT PROGRAMMES MEANS THAT APPLICABLE STANDARDS, REGULATIONS AND GUIDANCE MAY SHIFT DURING THE DEVELOPMENT PROCESS.”
Similarly, it is important to establish which standards and guidance will be applied. The drug delivery device regulatory framework is constantly changing and the long-running nature of device development programmes means that applicable standards, regulations and guidance may shift during the development process. Examples include updates to ISO 11608 for injection systems, ISO 10993 for biocompatibility, US FDA guidance for “five nines” reliability and, more recently, Essential Drug Delivery Outputs (EDDOs).
The implication of changing regulatory standards on the industrialisation phase is that strategies and requirements for design verification and validation (V&V), stability programmes and clinical studies can evolve during development. This can strongly influence the requirements for pre-launch supply of components and devices and, therefore, the specifications and capabilities of manufacturing systems, such as tooling, assembly and inspection equipment. These are long lead-time items where specification or implementation changes mid-stream can have major implications.
UNDERSTAND AND DOCUMENT THE DRUG DELIVERY SYSTEM DESIGN
Having a strong understanding of the device’s performance – its sensitivities and the limits of the design envelope – is highly recommended (Figure 1). Without it, characterisation, optimisation, implementation and troubleshooting become inefficient and problematic.
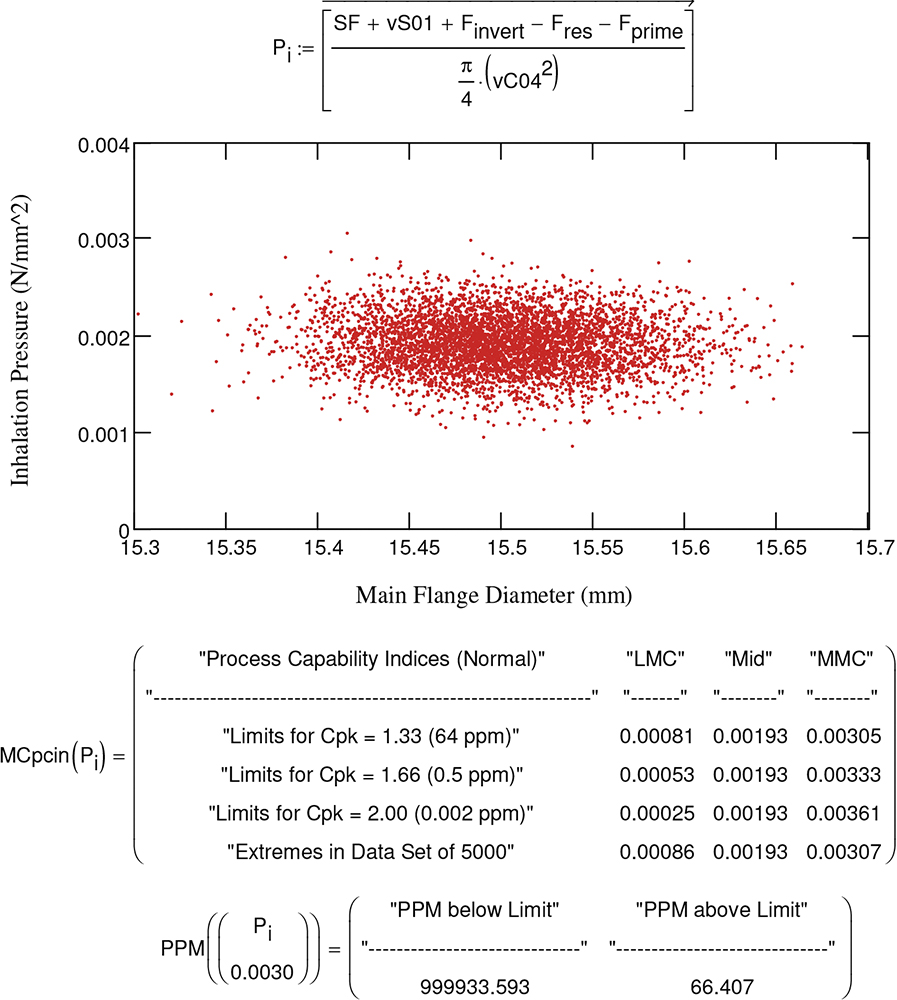
Figure 1: Sensitivity analysis: one tool to help establish and demonstrate design understanding.
For a DDS, this understanding and the documentation of it becomes a regulatory imperative. For example:
- The European Medical Device Regulation, which has increased focus on the device constituent part compared with the Medical Device Directive, requires that manufacturers ensure that all general safety and performance requirements are demonstrated as being met, and that manufacturing processes are appropriate.
- The design transfer section of ISO 13485 stipulates a number of requirements for manufacturers, including ensuring that design outputs are suitable for manufacturing before becoming final production specifications and that production capability can meet product requirements.
- The requirements set out by the recent FDA EDDO guidance extend to the need for documented manufacturing control strategies that demonstrate that essential outputs are understood and fully controlled.
Insufficient planning of how to meet these requirements can significantly impact the industrialisation and scale-up phases of a development programme. Having to build the necessary levels of device understanding late in the day – for example, to support final optimisation, tooling qualification or specification of test and inspection regimes – is a common cause of inefficiency and delay.
WORK WITHIN INDUSTRY STANDARDS AND ACCEPTED NORMS, OR BE PREPARED WHEN YOU CANNOT
The drug delivery industry is built on many well established, standardised elements. These include drug product primary packs, such as syringes, cartridges, vials, stoppers, needles, pressurised metered dose inhaler (pMDI) canisters and powder capsules, as well as materials, filling processes, etc. This standardisation brings benefits such as consistency, interchangeability and efficiency, but it does have a downside – device or technology innovation programmes that rely on making changes to any of these standards and norms can become more difficult.
In cases where these constraints can stifle innovation and result in suboptimal solutions for patients, there may be a strong argument for pushing back on them. When taking this route, it is important to go into the development programme with eyes wide open. It is possible to develop concepts and prototypes using non-standard elements, and doing so can present new opportunities.
However, engagement with CMOs and the supply chain at the start of industrialisation is not the time to discover that you are trying to swim against the flow. Establish this early on in the programme and make appropriate adjustments.
ADOPT A SYSTEMS ENGINEERING APPROACH
Drug delivery systems such as autoinjectors and pMDIs may seem relatively simple compared with large, complex systems. However, taking a systems engineering approach from the outset is still strongly recommended, as it can yield benefits and prove particularly helpful at avoiding – or resolving – issues that arise during industrialisation and scale-up. Such benefits include the use of sub-system verification, which can support efficient troubleshooting of late-stage issues, and the involvement of all disciplines throughout development to help prevent late-stage issues arising in the first place due to a perspective being overlooked early on.
Primary Pack Functionality
Consideration of sub-systems, interfaces and interdependencies is especially important for DDSs that incorporate existing primary packs that must meet additional levels of functionality beyond their original requirements. For example:

Figure 2: Primary packs as sub-system elements.
- A breath-actuated inhaler that relies on attributes of the pMDI canister (e.g. geometry and tolerances, force characteristics through life, valve opening characteristics)
- An autoinjector that relies on attributes of a prefilled syringe, such as geometry and tolerance to control timing of needle cover release, glide force to avoid excessive delivery times and glass robustness to avoid impact breakage.
These primary packs were not originally designed to be part of such systems, and hence are not optimised or controlled for this purpose. Suppliers will not necessarily be willing – or able – to hold and/or adapt key quality attributes to meet specific device needs (Figure 2).
User/Drug Interaction
For novel DDSs, the user/drug interaction is assessed through clinical trials. Early studies check for safety and initial efficacy, with some flexibility on which device is used. In Phase IIb and especially Phase III, however, where dosing and delivery performance is finalised, devices need to be more closely representative of the commercial product. This means that the industrialisation phase is often concentrated between pilot stage verification and the Phase III study. Delays to these stages bring a very heavy cost; therefore, it is critical to get the next two interactions right.
Drug/Device Interaction
Early demonstration of drug/device interaction is often carried out on development formulations, or surrogate liquids or powders, in prototype or pilot devices. Availability of representative drug can be either impossible or heavily limited due to cost.
Design verification through performance testing of a truly representative combination of drug and device can only occur following the industrialisation stage, when commercial production systems are approaching final qualification. If there are sensitivities in performance (e.g. delivered dose, particle size distribution, injection time, injection depth), this is not a good time to find out.
To mitigate this risk, development work should identify and investigate potential sources of variation. These can include temperature (viscous drugs), settling over time (suspensions), contact material and environment (drug stability), formulation robustness to delivery conditions (high stresses, due to pressure or vibration, e.g. ultrasonics), moisture ingress (dry powders) and electrostatics (powders and fine mists). Implications for system elements such as power sources, needle gauges, airflow characteristics and powder handling can then be resolved.
For platform systems, the full range of formulations intended for delivery needs to be considered when capturing input requirements. Decisions on what to include and what to exclude may be difficult but are necessary.
User/Device Interaction
Many DDSs rely on interaction with naïve patients and, therefore, need to be robust to variation. For example, it is advisable to base a device’s user interface on users’ pre-existing mental models and the different ways in which they may interact with the device. This emphasises the importance of good human factors/usability engineering (HF/UE), but also of applying good engineering design principles.
It is advantageous for the developer that these interactions can be investigated and de-risked well before the industrialisation phase. User populations are known and prototype devices of a reasonably high fidelity can usually be sufficient to support assessment through HF/UE studies and expert review.
The user/device interaction needs to be considered in both directions, however. Not only must the user be able to successfully prepare and operate the device, but the device itself must be able to withstand interaction with the user, such as gripping, dropping and foreseeable abuse. It is during industrialisation that device features critical to success are finally fully representative (e.g. snaps, clips, wall thicknesses, mass, material properties) and that units are available in large enough quantities for sizeable test programmes and actual use studies to be carried out.
“RISK MANAGEMENT, ENGINEERING ANALYSIS AND TESTING IN EARLIER PHASES MUST BE APPLIED RIGOROUSLY TO ENSURE AS FAR AS POSSIBLE THAT UNIDENTIFIED POTENTIAL FAILURE MODES DO NOT PRESENT THEMSELVES AT LATE STAGES OF DEVELOPMENT.”
Risk management, engineering analysis and testing in earlier phases must be applied rigorously to ensure as far as possible that unidentified potential failure modes do not present themselves at late stages of development.
CONSIDER THE ENTIRE DRUG DELIVERY DEVICE ‘ECOSYSTEM’
For medical products such as a DDS, the “device” element extends beyond the core device itself, to include items such as labels, instructions for use and tertiary packaging, as well as any supporting applications or other digital assets (Figure 3).
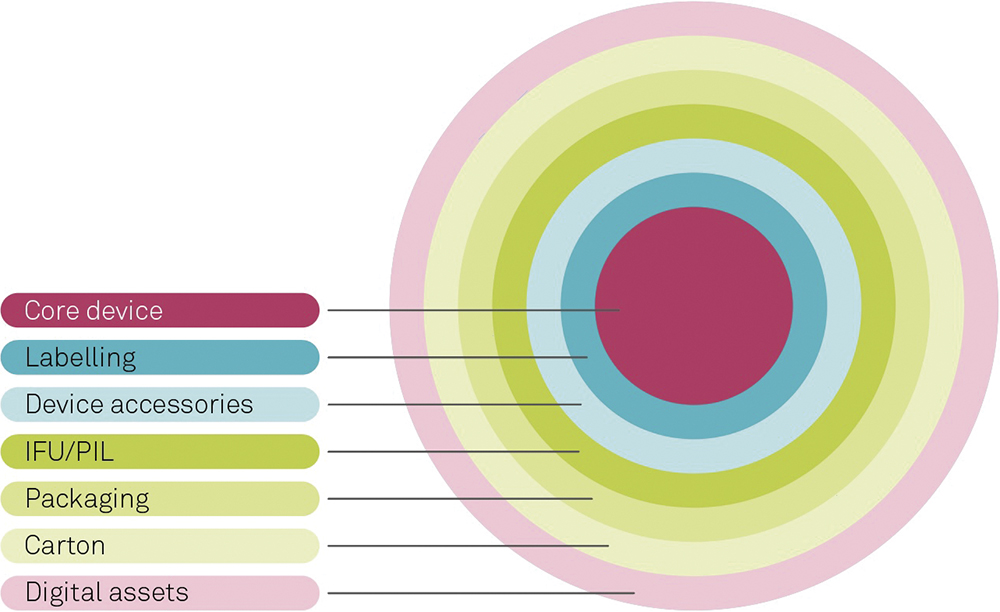
Figure 3: The broader device system.
Final design V&V of the DDS must be carried out on commercially representative versions of all these elements. However, experience shows that some are not given the necessary attention early enough in the development, or with sufficient focus. This can lead to delays, such as to design verification testing or summative HF/UE studies or – at best – extra loading and stress into what is often already an intense and demanding phase of work.
All elements of the DDS need to be progressed in parallel. It is bad enough for the device to find itself on the critical path without it being due to delays in the availability of, for example, labelling equipment or carton inserts.
Another good reason for parallel development of the entire system is that it can facilitate the incorporation of features into the device design, which can then be referred to, directly or indirectly, such as in the instructions for use (“remove the blue cap”). This can help to ensure that the overall package (including packaging) is optimised to support safe, effective and intuitive use. Leaving development of the ecosystem until later can close the doors on potential easy wins.
BASE DEVELOPMENT AROUND ROBUST RISK MANAGEMENT ACTIVITIES
Some DDSs have higher risk profiles than others, but there is always an imperative to ensure safe and effective use. Starting early with rigorous risk management activities, combined with an effective HF/UE programme, ensures that this can be demonstrated during design V&V.
The path to achieving low residual risk should become clear during detailed design, based on optimisation of the design and specification of production systems and control strategy. Any need for late-stage risk reduction, such as through design modification or additional inspection/test measures, can have a significant adverse impact on industrialisation. This should be a phase featuring implementation of known risk mitigation measures, not identification of new ones.
CAREFULLY PLAN AND MANAGE THE SHIFT FROM DEVELOPMENT TO MANUFACTURING ORGANISATIONS
The tools, processes and mindsets present in organisations specialising in rapid design development often differ from those in commercial manufacturing. Both are appropriate, and CMO partners are ideally involved in development during detailed design phases so that collaboration can start early. However, significant transfer of activities and responsibilities between organisations usually occurs during the industrialisation phase, and this can be a source of challenge.
“EFFECTIVE COLLABORATION ACROSS FUNCTIONS IS CRITICAL THROUGHOUT DDS DEVELOPMENT PROGRAMMES, BUT STAKES AND RISKS ARE OFTEN HIGHEST DURING THE INDUSTRIALISATION PHASE.”
Systems being transferred need to be compatible, decision-making processes need to be clear and effective and the collaboration in previous phases needs to have ensured joint buy-in and “ownership” of the design. Careful selection of the right partners is critical, as is generation of effective transfer plans, including definition of roles and responsibilities. Effective collaboration across functions is critical throughout DDS development programmes, but the stakes and risks are often highest during the industrialisation phase.
MANAGING INDUSTRIALISATION OF DRUG DELIVERY SYSTEMS
One of the challenges with DDSs is that delays at the industrialisation phase can be extremely costly. Late-stage programme milestones often drive critical activities, including supply of devices for clinical trials, sign-off to build registration batches or filings to hit important dates, such as an exclusivity window.
There are many potential causes of delay during industrialisation or scale-up, including poor planning, resourcing challenges and contractual negotiations with supply partners. Another is the need to resolve late-stage technical issues that are preventing performance requirements being met successfully. Specific challenges require specific mitigations, but a good general rule of thumb is to allow for as much “headroom” as possible while remaining commercially competitive. Examples include:
- Not specifying over-constraining or over-optimistic input requirements – do not turn “wants” into “musts” or “shoulds” into “shalls” if not necessary
- Understanding the performance window and keeping away from the edges, especially early on, to avoid problems arising later on
- Being more pessimistic than optimistic in working assumptions, such as for engineering analyses or worst-case test conditions
- Building in as much clearance as practicable between the process capability at which the design meets requirements and the process capabilities achievable from manufacturing systems, typically featuring high cavitation tooling and automated assembly
- Avoiding any processes that are on a knife-edge or highly sensitive to input variables
- Avoiding concepts that deviate from established principles and technologies or that depend on multiple levels of innovation, such as having more than one core technology that is at a low technology readiness level.
SUMMARY
While all DDS development programmes are different, bringing their own challenges and opportunities, over time, it becomes clear that some late-stage issues occur more often than others. Anticipating them early, or putting measures in place to address them efficiently when they do arise, can help to ensure a path to successful industrialisation.


